AU: 5-31-2017:
Results of OAADPr experiment. Results for 2xE0 will be uploaded soon.OAADPr results.pptx
AU: 5-23-2017
Attention: Using XG-batch 2 Sirt3-While I was looking at the R1, R2 experiments I did, I realized that there are extra peaks in the chromatogram. The two peaks are other than the substrate and product peaks. Their retention times are ~11 and 12 mins and their peak area is increasing with time. Since this wasn’t affecting the product peak quantification, it did not occur to me as a concern. But I revisited the data and found that the total area of product + substrate is also reduced.
As of now, I do not know why this happened. I ran same peptide (600 uM, same batch) I did not see these extra peaks. I ran same amount of enzyme (XG-EB2) as a control, and did not see these extra peaks. These extra peaks are not present when I use XG-EB 1. I am attaching a powerpoint which includes extra peak information. One possible explanation could be that the protein preparation is cleaving the substrate peptide over the time. This batch may have some protease contamination.
extra peaks XG-EB II.pptx
RC: Revised plan of expts. Revisions have been made to the version sent by email, so use this version. Dates need to be added bearing in mind deadline.
Updated- New Experiments 5-2-2017 RC comments.docx
Additional details regarding the preliminary validation steps:
a) in the draft schedule above (part 2), I've mentioned that a vmax experiment (3000uM NAD, 0 NAM, 0 HKL) can be used to validate the consistency of enzyme batches.
I would suggest doing the following for a rigorous validation:
-3000 NAD, 0 NAM, 0 HKL: use the full set of time points 0-120 (once, no duplicate), calculate the initial rate, then calculate the ratio of the old batch and new batch initial rates under these conditions (call this ratio R1)
-500 NAD, 0 NAM, 0 HKL (or some other NAD where we have full set of time points in previous expts): use the full set of time points 0-120 (once, no duplicate), calculate the initial rate,
then calculate the ratio of the old batch and new batch initial rates under these conditions (call this ratio R2)
-Then compare R1 and R2. If the enzymes have the same kinetic properties, but only different E0s, then the ratios should be the same because the rates are proportional to E0.
-Do the above for two batches of enzyme
-Choose the batch that has closer values of R1 and R2 as the batch to use for all subsequent experiments because this is the batch with more similar kinetic properties.
Use the value of E0 calculated here to align the initial rates between old and new batches.
Note: this is essentially equivalent to a vmax and Km calculation. The slope of the reciprocal plot will be Km/vmax and the condition that R1 and R2 are sufficiently close
is equivalent to Km's from the old and new batches being sufficiently close. Once this condition is validated, we make the approximation that Km and kcat are the same for the old and new batches,
and then we have Km/kcat*E0,old for the slope of the old batch and Km/kcat*E0,new for the slope of the new batch. Then, the ratios of the slopes of the batches gives us E0,old/E0,new which
is the alignment factor by which new batch initial rates should be multiplied so as to allow combination with data from the old batch.
Here, E0 refers to the concentration of active enzyme -- not necessarily same as the total amount of enzyme (some of which may not be active) obtained from Bradford assay.
since we require that the Kms are close to each other above, if this is validated, the kcats should then also be close and under these assumptions we can calculate
ratio of active enzyme concentrations. We will also make note of the total enzyme concentrations from Bradford or 280 measurement and compare.
b) having chosen an enzyme batch, I would then suggest one further preliminary study to assess the accuracy of XG's initial rate calculations in the presence of HKL. (As you know,
we are encountering some issues with accurate initial rate calculations in the presence of HKL.)
-Choose one of the experiments under part 2 of the schedule above (200 HKL). Measure the full set of time points 0-120 (once, no duplicate) and calculate the initial rate.
-Repeat the experiment --with 0.5x or 2x the amount of enzyme-- (0.5E0 or 2xE0). As you know, the initial rate should then be 0.5x or 2x the original initial rate.
If it does not, your method of initial rate calculation may have a problem.
-Find a method for calculating the initial rate that properly scales by 0.5x or 2x as expected.
Note that this experiment can be viewed as a "duplicate" experiment because all we are doing is changing the amount of enzyme in a known way.
This experiment will also help us assess the effect of minor changes in buffer composition due to use of different amounts of undiluted enzyme.
c) In the meantime, XG will be studying methods for obtaining more accurate initial rate values using our old data (given the issues encountered with IR calculations in presence of HKL).
Based on all the above, we will be able to setttle on the best plan for remaining experiments on the schedule (part 2). In particular, since
we then know how to calculate initial rates most accurately, we will be able to decide whether to do 0-40 mins in duplicate,
or 0-120 mins without duplication (in the event the full set of times
is preferred for accurate IR calculation) for the remaining experiments in part 2 of the schedule.
The draft schedule can be finalized after we finish the above and analyze the results (in a couple of days if needed).
The plan for dose response (part 1) will not change.
XG, should you like to discuss, please let me know.
For our info, we should know how much urea enzyme is still with 2bind.
AU: 4-11-2017: Schedule and plan for t-sirt3 initial rate experiments-
Plan for truncated Sirt3 initial rate experiments.docxRC (4/6): Some notes about planning of final IR expts:
Planning complicated by limited material, otherwise would be much easier. We already have most data we need, but we might as well finish up enzyme.This has not been the highest priority.
Considerations:
-Any objections from referees to lack of some duplication?
-One data point at 15000,15000 did not fit well.
-We may want to do some low NAD (e.g., 3000uM), high NAM expts where the model predicts rates w,w/o HKL are close
Options:
1) Duplicate w, wo HKL at 15000,15000
And one w, wo HKL at
3000 nad, 12000 NAM
2) Or only latter w duplication.
3) Or two w,wo HKL at
3000 NAD, 7000 and 12000 NAM
(Up to 3000 tried so far)
--Bear in mind that 3000,12 NAM may be noisy:
-At 3000,200 NAM and 200 HKL (inhibition), % cv is 7%
Rate is 0.123
-Predicted rate at 3000,12000 0 HKL
is .0359
Cv may be 25% (very roughly)
-For comp 15000,15000 should be around 0.06-0.07
Cv may be 15%
--So Alok can start asap, should we carry out remaining reactions in two stages? First 3000 NAD, 12000 NAM (w, wo HKL), then choose last expt thereafter
He could start this rxn while final expt planning done. What would be the rough schedule (for rxns, HPLC analysis etc)?
But provide your thoughts on options above/reply to question about duplication above, first.
RC (4/7): After providing your comments: Please provide estimated schedule if remaining expts are done in two stages as mentioned above (3000uM NAD/12000uM NAM first). After fitting second expt could then either be 3000/12000 or 15000/15000 repeat.
Also do comment on whether you anticipate 3000/12000 to be highly noisy and if you would suggest 3000/7000 instead (bear i mind my considerations for expts at top above).
AU: 4-5-2017 Updated-
HPLC method-kinetics.doc
AU: 3-28-2017
HPLC method-kinetics.docHPLC assay protocol-
AU: 3-23-2017 Extra high NAM-NAD experiment schedule.
More High NAD-NAM plan -1.docx
AU: 12-22-2016: Schedule updated to include Sirtainty assay.Re-Updated 12-22-2016-Task Alok.doc
AU: 12-15-2016: Updated schedule- Re-Updated 12-15-2016-Task Alok.doc
==
I have not prepared detailed Sirtainty schedule as this will the first time I will be doing this assay. After discussion with XG, I know that the I have enough enzyme. After Sirtainty assay, no enzyme will be left from the current batch.
I will upload detailed Sirtainty schedule in couple of days.
Thank you,
==
AU: 12-9-2016: NAM experiment updated.
Updated-NAM experiments_12.05.2016-12-9-2016.docx Sirt3 inhibition-NAM-K122-enpoint-Dixon-Update-12-9-2016.pptx
AU: 12-6-2016: Updated NAM-K122 experiments-
Updated-NAM experiments_12.05.2016.docx
AU: 12-5-2016; NAM studies and seubsequent email replies.Resposne on Recent Email_12.05.2016_update.docx
Resposne on Recent Email_12.02.2016_update.docx
AU: 12-1-2016: NAM titration and some endpoint assay on K122-MnSOD
NAM titration and endpoint assay-K122-12-1-2016.docx
Email reply/discussion:
Email from Raj 11-30-2016.docx
I will start working on this schedule.
NAM experiment on page 4 of this revised schedule.
Re-Updated 11-18-2016-Task Alok.doc
AU: 10-14-2016: Raw data 50 and 100 uM NAD initial rate- This table will be updated as data comes in. The finala analysis will be done once all data points are available to calculate MM constants.
Initial rate data.xlsx
AU: 9-28-2016: schedule updated again-
Re-Updated 9-28-2016-Task Alok.doc
AU: 9-26-2016: Updated schedule. Updated 9-26-2016-Task Alok.doc
AU:9-23-2016
Initially we plan to do initial rate reactions one by one e.g. make a reaction mix incubate for specified period of time e.g. 10 min, them do another reaction mix and carry out reaction for another time point and so on.
In initial rate reactions, there are 6 time points (10, 20, 30, 40, 80 and 120 min, for Km NAD determination) for each NAD concentration.
I am testing, if I can combine all these reactions and take an 50 uL aliquote at specified time points and quench the reaction with TFA.
I tried with 25 uM NAD + 600 uM K122 and result is very similar to previous data. The % product formation is comparable 0.66 % vs 0.63 %.
The only difference is amount of absolute pmoles formed. Since in this method, the total reaction mix (50 uL) being diluted by extra 5 uL which can be adjusted if needed.
I will upload the data once it is ready.
AU:9-21-2016; Sirt3 reaction in presence of 25 uM NAD, 600 uM K122 and 5 uM Honokiol- 25 µM NAD - 600 µM K122-MnSOD peptide.pptx
AU: 9-20-2016
Updated with dose response curve-Reactions with 350 uM Honokiol.pptx
Reaction with 350 uM Honokiol- Reactions with 350 uM Honokiol.pptx
AU: 9-15-2016: repeat experiment (50 uM NAD + 600 uM K122- peptide in presence of 5 uM Honokiol)
5 uM repeat experiment part of AU49.pptx
=========
AU: 9-14-2016: Information about Carba-NAD and Thioalkylimidate
Structure and synthesis in in this paper-szczepankiewicz 2012-carba NAD.pdf
I will send request to companies, which we have used in the past, and ask them if they can synthesize it.
=
AU: 8-30-2016: Format updated AU48-PMC-AU3
Final data-Experiment AU48-PMC-AU3.pptx
AU: 8-24-2016: AU48-PMC-AU3:
Email conversation started on 08-19-2016.docx
Final data-Experiment AU48-PMC-AU3.pptx, AU48-PMC-AU3.xlsx
AU:8.23.2016Final data-Experiment AU48-PMC-AU3_update.pptx
AU: 8-17-2016: AU47-PMC-AU2:
AU-47-PMC-AU2b.xlsx, AU47-PMC-AU2a-b.pptx
AU: 8-15-2016:
"Deacetylation of MnSOD by PARP-regulated SIRT3 protects retinal capillary endothelial cells from hyperglycemia-induced damage"
gao-2015-Sirt3-PARP.pdf
endothelial cells from hyperglycemia-induced damage
AU: 8-12-2016: Data for AU46-PMC-AU1:
Experiment AU46-PMC-AU1.pptx, AU46-PMC-AU1-Day 1-2-Final data.xlsx
RC: Comments regarding both AU and XG experiment 1
au xg minimal substrate expt comments 8-14.docx
AU: 8.9.2016: Please see updated schedule.
Updated 8-9-2016-Task Alok.doc, Response to remaining questions_8.9.2016.docx (response to remaining questions may be uploaded by XG)
AU: 7-28-2016:
CRO qotes for HPLC experiment:
1: Avomeen- PMC072716k.pdf
AU: 7-11-2016: Today's experiment- Results expected tomorrow (7-12-2016)
10: Assay with Different concentration of Honokiol
Enzo 5U/Rxn for 30 min. Range of Honokiol will be used:
10, 50, 100, 200 uM
2mM NAD, and 10 uM K-122 MnSOD
10a) with DMSO and with Honokiol (6 Rxn)
100 uM NAD, 600 uM K122 MnSOD
10b) with DMSO and with Honokiol (6 Rxn)
AU: 7-8-2016: Experiment AU44- estimation of saturating K122-MnSOD peptide concentration- AU44-Sirt3-K122-MnSOD-Saturation.pptx
AU:7-8-2016: Experiment AU43-Activity of Sirt3 on K122-MnSOD peptide.AU43-Sirt3-K122-MnSOD.pptx
AU: 7-7-2016 Updated schedule- Task Schedule-Alok 7-7-2016.doc
I removed without DMSO initial rate. This can be done later. I will plan to do an experiment to make sure 600 uM is saturating papetide before I proceed further (or I can make it 1 mM).
==
Your nonsaturating nad screening still says 5 or 10 uM peptide. Pls fix
AU: Fixed
RC: This has only been fixed in one place. Other still says unsaturating NAD with 5 and 10uM K122 peptide.
It will be advisable to do saturating peptide initial rate expts (no honokiol) before saturating nad. This change should be made.
Indicate whether you are planning to do 11,12 in parallel.
Also I need to see completion date for each of the subtasks in 11,12 asap.
=
AU: Task 11 and 12:Initial rate with 5% DMSO (K-122-MnSOD and in-house enzyme)
11: Initial rate with saturating peptide (42 rxn)
11a: 600 uM K-122 MnSOD (completion date: 7-20-2016)
100, 200, 400, 600, 800, 1200, 2000 uM NAD
Time: 5, 10, 20, 40, 80, 160 min
11b: with 5% DMSO (42 rxn) (completion date 7-29-2016)
600 uM K-122 MnSOD
100, 200, 400, 600, 800, 1200, 2000 uM NAD
Time: 5, 10, 20, 40, 80, 160 min
RC: You need to give some indication of how you determined that 600uM was saturating.
AU: Since we don't know the Km of K122-MnSOD peptide, it was just a guess that 600 uM will be saturating concentration. I don't think the Km of this peptide go beyond 100 uM. As long as Km is below 120 uM, 600 uM should be saturating. After doing the initial rate, if the data doesn't fits well, this can be modified and refined.
RC: After doing what initial rate? We don't want to redo several initial rate experiments in the planned series because the concentration was not saturating.
You should quickly verify with minimal expts whether 600uM is saturating.
AU: 7-8-2016: updated schedule to include this experiment. Task Schedule-Alok 7-8-2016.doc
12: Initial rate ; saturating NAD
12a: 2 mM NAD (42 rxn) (Completion date 8-9-2016)
25, 50, 100, 300, 500, 600, 1000 uM peptide
Time: 5, 10, 20, 40, 80, 160 min
12b: 2 mM NAD with 5% DMSO (42 rxn) (Completion date 8-14-2016)
25, 50, 100, 300, 500, 600, 1000 uM peptide
Time: 5, 10, 20, 40, 80, 160 min
See below - now you might do w/, w/o honokiol in parallel instead.
Should 0% DMSO be done after w/, w/o honokiol in 5% DMSO?
AU: I can move honokiol experiment up before #11 (keep DMSO experiment for later).
I will upload the detailed schedule end of the day.
RC: Before 11 or before 12? Keep 0% DMSO for later?
W/o honokiol cannot be done without DMSO. The appropriate control is with DMSO.
==
AU: Before #11, #13 can be done- (All the reactions below have 5% DMSO)
13: Initial rate with Honokiol (in 5% DMSO)
13a: Initial rate with saturating peptide
600 uM K-122 MnSOD
100, 200, 400, 600, 800, 1200, 2000 uM NAD
Time: 5, 10, 20, 40, 80, 160 min
RC: ?? What is the difference between 11b and 13a?
Hence my questions about "before 11".
Please clean it up.
13b: with Honokiol (concentration of HNK TBD, based on #10)
600 uM K-122 MnSOD
100, 200, 400, 600, 800, 1200, 2000 uM NAD
Time: 5, 10, 20, 40, 80, 160 min
14: Initial rate ; saturating NAD (84 rxn)
14a: Initial rate with saturating NAD
2 mM NAD
25, 50, 100, 300, 500, 600, 1000 uM peptide
Time: 5, 10, 20, 40, 80, 160 min
14b: with Honokiol (concentration of HNK TBD, based on #10)
2 mM NAD
25, 50, 100, 300, 500, 600, 1000 uM peptide
Time: 5, 10, 20, 40, 80, 160 min
==
Also it may be advisable to do initial rate in presence of honokiol and saturating peptide directly after the above (or in parallel). I.e. after saturating peptide initial rate experiments without honokiol).
Due to the fact that your schedule is pushing into late August.
We will decide after getting the screening results.
Please make a note of these points on wiki.
Also, it appears your schedule does not account for time for duplicate expts, which are needed for publication. When would those be done?
RC: Not answered.
AU: Yes, my schedule does not account for repeat experiments, as it will take long time to do. After initial rate, if we see something interesting, I can repeat that experiment immidiately.
FYI: If not in reasonable time, your data may not be publishable in an upcoming paper.
AU: What is the theme of the paper? can we do targated experiments?
RC: I would like to know how long the repeat initial rate expts for saturating peptide w/,w/o honokiol will take,
It appears you are planning only for interday duplication.
When will you determine whether XG will be using one HPLC concurrently with your expts?
AU: XG is also planning to do HPLC based initial studies so I will be left with one HPLC.
RC: I told XG that this needs to be assessed after receiving your proposed schedules assuming concurrent work.
Moreover all the initial rate study plans in presence of honokiol depend on the results of your screening studies.
AU: I am almost done with initial screening (#9). I will upload data tomorrow. This will give me conditions for reaction, not the honokiol concentration . The task #10 will decide what [honokiol] concentration should be used for the initial rate.
In addition, I would like to know asap what in-house enzyme preparation will be used for each set of expts.
AU: Starting from task #11, I will use in-house enzyme as mentioned earlier.
RC: I am asking both of you which batch of in-house enzyme. New method with truncated enzyme or the same AE method with urea on the longer construct.
Updated schedule: AU:7-1-2016-Task Schedule-Alok 6-3-2016.doc
-----------------------------------------------------------------------------------------------
AU: 6-29-2016: Tentative plan for MnSOD-K122 peptide reactions- (tentative completion date for #9: on or before 7-1-2016):
9: Assay of Sirt3 with MnSOD-K122 peptide, Enzo Sirt3 5U/Rxn
9a) Saturating NAD with 5 and 10 uM K-122 MnSOD
9b) Un-saturating NAD (100 uM) with 5 and 10uM K-122 MnSOD
If 5U/rxn doesn’t work, then move to-
Assay of Sirt3 with MnSOD-K122 peptide, Enzo Sirt3 10U/Rxn
9c) Saturating NAD with 5 and 10 uM K-122 MnSOD
9d) Un-saturating NAD (100 uM) with 5 and 10 uM K-122 MnSOD
A full schedule will be posted soon for following (below), it is already written, I need to change the dates.
10: Honokiol dose response, reaction condition based on task #9
11: initial rate with DMSO
12: Initial rate with Honokiol (concentration will depend upon #10)
===
From: Raj Chakrabarti
Sent: Wednesday, June 29, 2016 10:53 AM
To: Alok Upadhyay
Cc: Xiangying Guan
Subject: FW: [Wikispaces] pmc-at : Task List from Alok was edited by aupadhyay
Comments:
-- I see you have opted to do screening (including expts without honokiol) before initial rates.
This means you will need to verify the substrate concentrations chosen for screening with honokiol were sufficiently low after getting the Km of the peptide, later.
Yes, and my hope is the Km may be between 30-50. Going lower than 5 uM, regardless of Km, will not be accurately quantifiable with HPLC. Please suggest, If I should determine the Km first.
-- By "doesn't work" do you mean the activity is too high or too low?
It appears you mean too low since you will be increasing [enzyme] in that case.
If it is too low, then move to high Sirt3.
-- What are the reaction times? Will they be reduced if the activity is too high?
Reaction time I will be testing 5 min and 30 min. As of now I am not thinking going below 5 min. Higher side will be better. If I add modulator, there should be sufficient time to interact.
-- *For both MnSOD and AceCS2, as noted earlier, making both substrates nonsaturating should only be used
as a last resort. We want to study the two cases where one of the substrates is saturating.*
I will change the [peptide] to 50 and 250 uM in case where NAD is nonsaturating.
-- Why would you run nonsaturating NAD+ at higher [enzyme] after finding the activity was too low?
Its plan dependent upon previous exp, which can be removed later.
-- What about initial rate without DMSO or honokiol? Is that to be done last?
This can be moved either just after task 9 or after 10 depending upon the outcome of task 9.
-- See my multiple previous comments about [honokiol] in initial rate studies on wiki.
As noted there we may ulitimately do two nonzero concentrations (one may be 10uM as reported in Nat Comm, other higher). -- Initial rate expts will be done with in-house enzyme
Yes, thank you
==
You don't need to check Km of peptide first necessarily.But you'll need to verify later as noted.
nonsaturating nad should be coupled with saturating peptide everywhere if possible.
AU: OK.
What is the plan for initial rate expts with a) saturating Nad b) saturating peptide? You need to break these down. I assume b will be first?
AU: Please see the schedule above. I can plan to do saturating peptide first.
Given that you roughly know Km nad, I assume you and guan are confident that the nad concentration in nonsaturating Nad expts (screening) is sufficiently low with respect to the detection of activation per the analysis I discussed with guan.
AU: I am using 100 uM and hope this will be sufficiently low. XG can further comment on this.
As noted the comments/clarifications
should be posted on wiki.
==
AU: 6-29-2016: Optimization of Sirt3 activity on AceCS2 peptide:
Based on experiment AU41, the activity of Sirt3 is very high in following condtion-
[NAD+] = 2 mM
[AceCS2] = 1, 5, and 10 uM
Reaction time = 5 and 30 min at 37 degreeC
Enzo Sirt3 = 10U/reaction
In above specified condition, the activity is very high. In order to see appreciable amount of activation, the % product formation should be low. To achieve this goal, following modifications can be made-
-Amount of enxyme should be titrated (starting 1, 2, or 5 U/rxn)
-The reaction can be carried out at room temperature (25 degreeC) or at 30 degreeC
[AceCS2] 1 uM is too low for the HPLC (people have used this concentration with Mass spec), this concentration should be eliminated.
-a sub-saturating [NAD+] can be tried if this condition effectively lowers the product formation.
AU: 6-23-2016: Experiment AU41: Sirt3 reaction with saturating NAD (2 mM), with 1, 5, and 10 uM AceCS2 peptide for 5 and 30 min reaction at 37 degreeC.
AU41-Sirt3-AceCS2-Low conc.pptx
AU: 6-23-2016:
| In the lab, minimum [peptide]: FdL = 10 uM, p53 = 50 uM, AceCS2 = 50 uM: Minimum [NAD+] = 100 uM |
||||||
| Enzyme |
Peptide |
Km Peptide |
Km NAD |
Ref |
% of Km (peptide) |
% of Km (NAD) |
| Sirt3 (102-399) |
AceCS2-(638TRSGKacVMRRLLR) |
33 |
Lei Jin et al 2009 |
151 |
||
| Sirt3 (118-399) |
AceCS2-(638TRSGKacVMRRLLR) |
28.7 |
Lei Jin et al 2009 |
N/A |
||
| Sirt3 (102-399) |
AceCS2-(638TRSGKacVMRRLLR) |
600 uM |
Lei Jin et al 2009 |
16.6 |
||
| Sirt3 (118-399) |
AceCS2-(638TRSGKacVMRRLLR) |
598 uM |
Lei Jin et al 2009 |
N/A |
||
| Sirt3 (102-399) |
FdL |
640 uM |
done in the lab |
15.6 |
||
| Sirt3 (102-399) |
FdL |
32 |
31.25 |
|||
| Sirt3 (102-399) |
FdL |
2000 uM |
Enzo |
5 |
||
| Sirt1 |
p53-NH2-HLKSKKGQSTSRHK(K-Ac) LMFK-OH |
10.3±2.6 |
Kaeberlein et al, 2005 |
485 |
||
| Sirt1 |
FdL-p53 peptide |
87.6±19.7 |
Kaeberlein et al, 2005 |
11.4 |
||
| Sirt1 |
p53-NH2-HLKSKKGQSTSRHK(K-Ac) LMFK-OH |
132.5±33.9 |
Kaeberlein et al, 2005 |
75.4 |
||
| Sirt1 |
FdL-p53 peptide |
191.9±21.7 |
Kaeberlein et al, 2005 |
52 |
||
| Sirt1 |
p53 peptide |
25 |
Rye et al 2011 |
200 |
||
| Sirt1 |
p53 peptide |
34 |
Rye et al 2011 |
294 |
||
| Sirt1 |
p53 (p53tide) |
61 |
Blander et al 2005 |
82 |
||
| Sirt1 |
FdL |
64 |
Enzo |
15.6 |
||
| Sirt1 |
FdL |
558 |
Enzo |
17.9 |
AU: 6-22-2016: Some of the kinetic values from the literature for Sirt1 and Sirt3-
| Enzyme |
Km |
Ref |
Peptide |
| Sirt3 (102-399) |
33 |
Lei Jin et al 2009 |
AceCS2-(638TRSGKacVMRRLLR) |
| Sirt3 (118-399) |
28.7 |
Lei Jin et al 2009 |
AceCS2-(638TRSGKacVMRRLLR) |
| Sirt3 (102-399) |
NAD:600 uM |
Lei Jin et al 2009 |
Km NAD |
| Sirt3 (118-399) |
NAD:598 uM |
Lei Jin et al 2009 |
Km NAD |
| Sirt1 |
10.3±2.6 |
Kaeberlein et al, 2005 |
p53-NH2-HLKSKKGQSTSRHK(K-Ac) LMFK-OH |
| Sirt1 |
87.6±19.7 |
Kaeberlein et al, 2005 |
FdL-p53 peptide |
| Sirt1 |
132.5±33.9 |
Kaeberlein et al, 2005 |
Km NAD |
| Sirt1 |
191.9±21.7 |
Kaeberlein et al, 2005 |
Km NAD |
| Sirt1 |
25 |
Rye et al 2011 |
p53 peptide |
| Sirt1 |
NAD: 34 |
Rye et al 2011 |
Km NAD |
| mSirt3L (54-334) |
2.44 |
Lei Jin et al 2008 |
AceCS2 |
| mSirt3L (54-334) |
NAD: 280 |
Lei Jin et al 2008 |
Km NAD |
| Sirt1 |
61 |
Blander et al 2005 |
p53 (p53tide) |
| Sirt2 |
19±0.85 |
Teng et al 2015 |
H3K9 acetyl peptide |
Update: As determined in the lab, Km NAD = 640 uM, using FdL peptide and Enzo Sirt3.
AU: 6-16-2016
Revised schedule- Task Schedule-Alok 6-3-2016.doc
RC:
a) recall AceCS2 peptide is the preferred choice between p53 and AceCS2; please prioritize it for the
screening expts by doing the AceCS2 screening first
AU: 6-20-2016- I did initial screening with AseCS2 peptide (Experiment AU39, below). For next step, I will start with AseCS2 peptide first (#9 in the schedule).
RC: I am also referring to this week's experiments - do the AceCS2 expts first if they are not done concurrently.
b) Schedule does not account for
MnSOD peptide (esp K122): when is it coming in, and the priority for endpoint screening with it once it does,
before proceeding with initial rate expts, which may be done with that substrate (will choose between AceCS2 and that).
AU: 6-20-2016: Yes, as of now MnSOD peptides are not included in the schedule but once I receive the peptides, I will update the schedule (endpoint assay, before initial rate).
I just spoke to a rep from peptide synthesis company, she will let me know tomorrow exactly when the they will ship. As of now, most of them are in QC and here is the realtime information from the website.
MnSOD peptide update: I spoke to rep from the company, she said that peptides are in QC and once they pass QC, they will ship either on Tuesday or Wed next week.
RC: You should schedule them as soon as you know the date they are supposed to arrive. If there is some change later you can edit it again at that time.
I will wait for replies to my other questions.
AU: 6-21-2016: Updated email reply. The new information is in the red font. Email-Raj-2-6-20-2016.docx
AU: 6-21-2016 Updated with table-Email-Raj-2-6-20-2016-1.docx

I will post the data for experiment AU39 (task #6 and 7 of the revised schedule) by EOD. Data update Experiment AU39- AU39-Sirt3-AseCS2.pptx
AU: 6-10-2016: Experiment AU38 (Task #4-5 of the schedule)-
AU38.xlsx; AU38.pptx
AU: 6-3-2016: Draft schedule: Task Schedule-Alok 6-3-2016.doc.
MnSOD peptides are expected to be shipped in 2 weeks, we will have better idea on Tuesday (as per Zi).
RC: The following expts are not required at this time: jump to 4 immediately.
3: Repeat the experiment (#2) with p53 (30 min reaction)
3a: 100 uM NAD, 10 uM P53
W/O DMSO, with DMSO, with 10 uM Honokiol (3Rxn)
3b: 100 uM NAD, 50 uM P53
W/O DMSO, with DMSO, with 10 uM Honokiol (3Rxn)
3c: 100 uM NAD, 100 uM P53
W/O DMSO, with DMSO, with 10 uM Honokiol (3Rxn)
RC: For the expts below, [honokiol] will depend on the results of endpoint screening.
9b: with 10 uM Honokiol
600 uM p53, 100, 200, 400, 600, 800, 1200, 2000 uM NAD
Time: 5, 10, 20, 40, 80, 160 min
9b: with 10 uM Honokiol
600 uM p53, 100, 200, 400, 600, 800, 1200, 2000 uM NAD
Time: 5, 10, 20, 40, 80, 160 min
RC (6/2): It not clear why none of the questions below were answered for 3 days. (Although it was stated that you should be able to proceed without requiring further
feedback, a revised schedule with certain imp dates and answers to several questions relevant to planning were asked for, and the revised schedule has still not been provided.
An update report was also requested last week and it was not provided.)
Answers to all questions and schedule should be provided, along with revised draft of schematics, by Fri.
Given the delay, the schedule becomes all the more imperative since MnSOD peptides may arrive before you make substantial progress with p53. In the schedule include the estimated date of arrival of MnSOD.
As noted I need estimated dates for some of the critical milestones mentioned.
Your figure updates are due today.
Productivity this week has been v limited.
Posting a few comments is far from being sufficient for a week's work.
RC (5/30): Since there was no report, I am also giving a brief outline as feedback:
-- enzo bulk enzyme is meant for screening not initial rate/mechanistic studies
Should we want to use higher activity enzyme, we could just use in-house without urea purification steps. We did urea purification to get higher purity.
Need to know whether we have enough in-house enzyme for both series of initial rate experiments.
AU: 6-2-2016: There is enough enzyme for me to do initial rate but not enough if me and XG use at same time. SM needs to purify more for enzyme.
substrate: p53 (until MnSOD arrives)
Reproducibility: are you referring at all to batch to batch variability? what is the plan for combining batches? settle this internally and confirm here.
AU: 6-2-2016: The current batches can be combined but the only issue as SM mentioned and we are aware of is the freez thaw cycle. All the enzymes are in -80 (individual batches) and in order to combine them he needs to thaw them and in this process we may see some reduction in activity.
RC: The limited amount of enzyme was not mentioned earlier this week.
Sudipto should explain how long it would take him to get the purified enzyme in the schedule on his page. Bear in mind that we do not want Sudipto to be delayed much more than a few days in testing the new purification protocol.
How do you plan to compare the results between batches if you don't characterize them or combine them?
It may be ok to use one (potentially combined) batch for honokiol expts and another for DHP experiments as long as we don't cross compare.
But then the MM studies in absence of modulator would need to be repeated.
Work out the plan. Of course, it is not my responsibility to advise on such routine scheduling details such as purification every time. You all should ensure that you
flag such issues in the future and dealt with them yourselves.
SM: Problems with combining enzyme batches and possible solutions (contributions from XG, AU and SM):
Problem with using different enzyme batches:
- In XG's experience, there is batch to batch variation in measured kinetic parameters
- identical experiments with different enzyme batches have yielded different results
- same problem has been observed in case of both in-house enzyme and Enzo enzyme, pointing to the need to have a large uniform batch of enzyme instead of many small batches
Possible solution to the above problem:
- combining different enzyme batches to have a single homogeneous batch
Problem with the above possible solution:
- Upon purification, fresh enzyme batches are aliquoted and stored in -80C.
- When required, an aliquot is taken out, thawed on ice and used.
- However, refreezing the unused sample is not recommended since it could lead to denaturation and loss of activity
- in order to combine batches, all the aliquots have to be thawed, combined and then re-frozen, which could lead to loss of activity (and all of the active protein), and is potentially very risky.
Possible solution to test if freeze thawing would affect activity:
- Take an aliquot each from 2 separate batches, thaw, determine activity
- Freeze unused sample, thaw again, measure activity again.
- Perform identical experiments in duplicate
- Major change in activity between enzyme samples freshly thawed and re-frozen/thawed would indicate loss of activity upon re-freezing and thus it would not be a good idea to combine the different batches.
- If no major change in activity, then the different batch aliquots could be combined.
RC: Ok -- as I mentioned, it's your collective responsibility to resolve this. Work it out and post the solution and schedule for Sudipto's purification efforts, bearing in mind that his work with new protocol should not be significantly delayed as mentioned.
AU: 6-9-2016: I did the reaction with following condition-
500 uM NAD+
250 uM pep1 (p53) peptide
37 degreeC for 30 min
1: Two samples, 1.6 U/ul and 0.5 U/ul were thawed from -80 and reaction was done. The enzymes were frozen again in -80 for 1 hrs and thawed again and activity was done (one freez-thaw cycle). Based on the data it appears that in this specified condition, the activity did not change, at-least for 0.5U/ul enzyme. There was problem in one of the reaction with 1.6U/ul (see reaction 2 in ppt) so it was incoclusive. We all agreed that, it is safe to combine batches. It would be good to have as many batches as required and thaw and combine them togather.
AU37.xlsx Experiment AU37.pptx
-- ideally do saturating peptide initial rate studies before saturating NAD+
AU: 6-2-2016: OK, I will plan accordingly. Is there any specific reason to do initial rate with saturating peptide first?
RC: Yes, because it is the condition tried in Nat Commun.
AU: 6-3-2016: Non-saturating NAD+ for sure as they mentioned it, but not sure about peptide. They say 0.5 ug of MnSOD but did not specify the volume of of the reaction. We can estimate the MnSOD concentration by estimating the volume e.g. this table was posted before-

If I assume the volume was 10 or 20 uL then the respective [MnSOD] will be 203 or 101 uM respectively. In a typical western blot reaction, the volume is generally 20 to 50 ul. This will correspond to range 40-101 uM MnSOD.
RC: You asked why I asked to do nonsaturating NAD+ first. We are talking about one substrate being saturating.
Not clear what you are getting at.
AU: 6-6-2016. The point I am making is, Nat comm paper may have used both substrate non-saturating.
RC: I believe we already covered this point in the past. It is not what you asked above and the subject of this thread -- you asked why we were doing saturating peptide first rather than saturating NAD+ first.
You should be able to roughly determine saturating [peptide] in advance of initial rate studies; this should not require initial rate studies (if this is not the case please elaborate but do not wait for a reply)
As noted you need to determine effect of 10uM honokiol on activity with p53 substrate in endpoint assay first (similar conditions to what you have sampled in the past with FdL substrate; start with endpoint study on saturating peptide, nonsaturating nad+; also saturating nad+, nonsaturating peptide)
AU: 6-2-2016: The FdL experiment was done in non saturating concentrations of both NAD and FdL peptide. Same experiment can be done with p53 peptide with 10 uM Honokiol and then I can do nonsaturating NAD and saturating peptide p53 and vice versa.
RC: Ok, please provide detailed list of these experiments and schedule. Also provide a brief written summary here of the results from the FdL peptide expts you mentioned -- what was the qualitative effect at 10uM and at what concentration did you start to see inhibition clearly. Keep it efficient and brief. For the saturating NAD+ initial rate study, e.g., the [honokiol] determination only requires endpoint assays at saturating NAD+. Same for saturating peptide initial rate studies. So plan accordingly.
If there is minimal effect on activity at 10uM use hplc on higher [honokiol] until we see an effect
AU: 6-2-2016: In doing so, I will be doing unorganized initial rate studies.
RC: I am talking about endpoint studies, of course, not initial rate studies.
AU: 6-3-2016: Q: My understanding of endpoint assay is- Enzyme reaction at substrate concentrations (and fixed time) where changing the substrate concentration doesn't change the product formation at a given time. The increase in product formation will take place only when time is changed. basically the enzyme will follow zero order kinetics. For practical purposes, the concentration should be saturating and time must be chosen in linear range. Because of this, I am struggling to define endpoint assay. A feedback on this will be helpful for the experimental plan.
This is what I had in mind when I wrote "..unorganized initial rate studies."
RC: I am talking about 10uM honokiol and the determination of [honokiol] to be subsequently used in initial rate studies. I am referring to exactly the type of screening experiments we did before. By endpoint we mean not carrying out a full set of measurements at successive times in order to obtain the time series of product concentrations. The single/final time at which we measure the product concentration can be anything we like. You need to discuss with XG -- I am using the term in the same sense as we have used it before when we used various concentrations of substrates and tested for EC1.5s, etc. XG, please make sure you and Alok communicate since these are minor semantic issues that I thought were clear.
Then run initial rate assays under these conditions
-- check whether binding of honokiol to SIRT3 mentioned in Nat Comm required substrates to cobind or whether binding occurred to apoenzyme
AU: 6-2-2016: Based on Fig9A (Pillai et al.,) the binding was with enzyme alone.
RC: Ok, this is an issue with their work since it does not necessarily mean honokiol can cobind with substrates.
-- need to know whether saturating nad+ series of expts could be run on old hplc concurrently with saturating peptide on new hplc
AU: 6-2-2016: Either of the experiments can be done on either HPLC.
RC: I am referring to running them concurrently. Also, you need to think about what expts you run on each.
I assume each set of initial rate expts (peptide or NAD+ saturating) will be done on one HPLC.
In the schedule indicate which HPLC is being used for which expts. and when.
-- there is also the issue of initial rate studies in presence of dmso only. see previously posted comments on this. discuss w guan and schedule as needed.
AU: 6-2-2016: After discussing with her, the experiment can be done but no concenssus on the what % of DMSO must be used, 1, or 5%. Or just treat DMSO as a "modulator" and do a four different concentrations of DMSO e.g. 1%, 5%, 10%, and 20% DMSO?
RC: No, you should use just one % DMSO -- a % that is sufficient to achieve a [honokiol] where you can see an endpoint effect. If 1% is sufficient, use that.
AU: 6-3-2016: On second thought, since DHPs can be soluble in only 5% DMSO, we should use 5% DMSO so it will cover both Honokiol and DHPs. If not I will use 1% DMSO as suggested,
-- If warranted we could do two [honokiol] in a mixed inhibition study, but need to understand hplc resource allocation first to determine how long this would take.
Should start with [honokiol] where we see an effect on endpoint activity, esp since with p53 we are not repeating a published expt (as with MnSOD)
Would both hplcs be available to you for initial rate studies. Discuss also with Sudipto vis-a-vis his schedule.
AU: 6-2-2016: As of now, XG is using old HPLC for couple of reactions, after that I can use both HPLC for initial rate. As I mentioned, one set at a time can be run on one HPLC.
-- need dates. Give date by which all initial rate studies would be done for p53
AU: 6-2-2016 I will update.
RC: Please do so by Fri.
-- Indicate whether other groups could use microarrays to screen for honokiol effects on many substrates in higher throughput – any potential issues? comment.
AU: 6-2-2016: I am not sure how microarray will be helpful in this regard. Could you please elaborate the idea behind using microarray.
The microarray will tell us if the modulator, in this case Honokiol, has any effect on global gene expression. Or we can have peptide library (~6000 peptide substrate on chip, the way it was published in another Nat communication paper, genome wide acetylone study) and see if Sirt3 can be activated in presence of Honokiol with different substrate. For this we have to be sure, at-least for one substrate, honokiol activates Sirt3.
RC: I mean the peptide library on a chip. It is true that we should see activation for one substrate first. The point is to see the substrate dependence (as in the case of resveratrol for the previous peptide on a chip paper).
-- the requested analysis regarding quantifiability of initial rate products with hplcs was not finished (for any substrate).
AU: 6-2-2016: Do you mean the minimum amount of non-acetylated peptide (peptide run alone) or run a actual reaction and see what is the minimum amount of product (deacetylated peptide) can be detected?
RC: I am referring to the use of whatever data you had in hand (including the FdL data of XG) to confirm that all the concentrations we usually run in initial rate studies can
be detected on both HPLCs for FdL substrate, p53 or both. You never drew any conclusions about this in a systematic report. We do not want any surprises, so we like
to see whatever analysis is possible in advance. See my comments immediately below.
FdL data of XG was meant to be used in that context; this was explained. Provide a summary of what conditions we can quantify in initial rate assays.
There was no (ppt) report posted on this topic. Only proposed initial rate experiments were posted. This issue of reporting has been covered in the past.
-- Need to look at literature to determine whether p53 is a physiologically relevant substrate of SIRT3 – provide links to relevant literature
AU: Here is one paper suggesting that p53 is substrate for Sirt3. Li et al 2010-p53-Sirt3.pdf. I will update if I find more papers.
RC: Please confirm that p53 was conclusively identified as substrate.
AU: 6-9-2016: It is not very clear that p53 is a Sirt3 target. They used p53 peptide to assess the deacetylate activity. Other authors show that p53 may bind to Sirt3.
Also please indicate whether there is evidence for physiological role of the other substrates you currently have in lab for SIRT3.
==================================
AU: 7-5-2016: Sirt3-MnSOD-p53 and literature:
See the paper "Mitichondrial sirt3 and heart disease" by Gupta and colleagues.
Gupta table 2 indicates p53 is sirt3 substrate.
AU: Table 2 of above paper refers to an article published in 2010 (Ref #73. Li S, Banck M, Mujtaba S, Zhou M-M, Sugrue MM, Walsh MJ. p53-induced growth arrest is regulated by the mitochondrial SirT3 deacetylase. PLoS One. 2010;5:e10486. doi:10.1371/journal.pone.0010486.[PMC free article] [PubMed] ), which we have already discussed on wiki around 6-9-2010. This was the only report which I could find suggesting that p53 may be Sirt3 substrate.
On the other hand it does not list mnsod as a substrate, although it is mentioned in the paper.
AU: This paper “"Mitichondrial sirt3 and heart disease: Cardiovasc Res. 2010 Nov 1; 88(2): 250–256 was published in 2010 whereas MnSOD papers came later… eg.
1: K122-MnSOD: Tao et al., Mol Cell. 2010 December 22; 40(6): 893–904. doi:10.1016/j.molcel.2010.12.013.
2: K122-MnSOD: Pillai et al., Nat Commun. 2015 Apr 14;6:6656. doi: 10.1038/ncomms7656.
3: K68-MnSOD: Chen Y, et al. 2011. EMBO Rep 12: 534
4: K53 and K59 of MnSOD: Qiu X, et al. 2010. Cell Metab 12: 662
“… although it is mentioned in the paper.”
AU: I think they have discussed MnSOD in the context of a pathway, and not as a Sirt3 substrate.
Please comment since you previously suggested p53 may not be acknowledged as a sirt3 substrate,
whereas we are doing experiments with mnsod as a substrate.
AU: Since there was only one paper which suggested that p53 may be a Sirt3 substrate, and the results are not conclusive.
Please post your comments in the relevant place on the wiki.
Raj
========================
RC (6.6.16): Ok, please make sure to answer my question on wiki regarding p53 as native SIRT3 substrate, and whether there is any evidence that the other substrates we have in house are more physiologically relevant, before those expts start.
XG (6.7.16): Three substrate peptides are currently available in PMC-AT lab (p53 peptide, H3K14 peptide, and AceCS2, details listed below).
*P53 peptide
Pep1: NH2-KKGQSTSRHK(KAc)LMFKTEG-COOH
Pep1c: NH2-KKGQSTSRHKKLMFKTEG-COOH
*H3 peptide
Pep2: NH2-ARTKQTARKSTGG(KAc)APRKQLC-COOH
Pep2c: NH2-ARTKQTARKSTGGKAPRKQLC-COOH
*K642acetylated human acetyl-coenzyme A synthetase 2 (AceCS2)
Pep3: NH2-TRSG(KAc)VMRRLLR-COOH
Pep3c: NH2-TRSGKVMRRLLR-COOH
In “The substrate specificity of sirtuins” published on Annu. Rev. Biochem. 2016, the in vivo substrate specificity of SIRT1/2/3 has been addressed (See Supplemental Table 2-4 below). P53 and H3K14 are SIRT1 substrates in vivo, and AceCS2 is SIRT3 in vivo peptide.
supp_the substrate specificity of sirtuins_2016_Bheda_SIRT1 sub.xlsx
supp_the substrate specificity of sirtuins_2016_Bheda_SIRT2 sub.xlsx
supp_the substrate specificity of sirtuins_2016_Bheda_SIRT3 sub.xlsx
RC (6.6.16): Also, depending on reply to my questions on whether Nat Commun claimed that MnSOD K122 peptide deacetylation was activated by honokiol, and depending on when MnSOD peptides come in, it may make sense to do some of the following expts for MnSOD K122 peptide as well:
|| Endpoint assays (two time points; 5 and 30 min, 10U/rxn)
4: Assay non-saturating NAD & saturating peptide
4a: 100 uM NAD, 600 uM p53, 1% DMSO (2rxn)
4b: 100 uM NAD, 600 uM p53, 10 uM Honokiol (2rxn)
5: Assay non-saturating peptide & saturating NAD
5a: 2 mM NAD, 50 uM p53, 1% DMSO (2rxn)
5b: 2 mM NAD, 50 uM p53, 10 uM Honokiol (2rxn)
||
| 6: Assay with Different concentration of Honokiol (with p53 peptide) Condition will be based on #4, but a range of Honokiol will be chosen e.g. 5, 10, 50, 100 uM |
before starting initial rate expts with p53 or other in house peptide (the claim is they are coming in 2 weeks and initial rate expts with p53 were scheduled around that time).
And, we need conclusive confirmation posted to wiki that all the initial rate experiments that have been planned will be accurately quantifiable on both HPLCs.
You and Alok should respond on these points on wiki. Beyond this the lab staff will need to collectively handle remaining details.
RC (6.7.16): If p53 is not a physiologically relevant substrate of SIRT3, it raises the serious question of whether demonstrating it is not activated by honokiol for this substrate is very relevant and whether we should do all our initial rate studies with AceCS2.
Hence, comment on the evidence regarding the relative activity of SIRT3 on p53 vs AceCS2, why we chose p53 for synthesis in the first place given that it was not generally mentioned as a SIRT3 substrate, and
XG (6.7.16): One of the reasons why we choose p53 was because p53-320 was the substrate deacetylated most efficiently by SIRT3, from among a panel of substrates patterned on p53, p65, histone H3 and Histone H4 acetylation sites (Based on Enzo user manual).
AU: 6-9-2016: Another reason we synthesized p53 peptide becasue we wanted to have panel of peptides so we can test if activator is substrate dependent or not. Also, if in future we work with other SIRTs then we have peptide ready in the lab.

RC (6.7.16): how much additional work/what changes to schedule would be required if we used AceCS2 instead.
XG (6.7.16): Endpoint experiments need to be done to compare either AceCS2 or p53 deacetylated more efficiently by in-house SIRT3. Proposed conditions
[In-house SIRT3]=10 ul
[AceCS2 or p53 peptide]= 50, 600 uM
[NAD+]=100, 2000 uM
Temp.= 37oc
Time point = 30 min
RC(6.12.16): Since Alok finished #4,5 with p53 I assume he would need to redo those with AceCS2 were we to use that.
XG(6.13.16): We can use AceCS2 to redo the #4 and 5. Alok is working on the schedule.
RC(6.12.16): I had also asked for an update on the MnSOD order. Please provide that so we can see where it will intersect with the schedule. See my previous notes on this and whether the timing (after AceCS2 endpoint screening, e.g.) would be such that we should do MnSOD K122 endpoint before doing initial rate studies.
XG(6.13.16): Based on Ziting’s peptide update, MnSOD order will be ready on 6.20.2016. It’s good the timing to finish AceCS2 and move on MnSOD.
RC(6.12.16): Do you see any reason not to use AceCS2 instead of p53 for SIRT3 HPLC experiments planned? I had asked whether this would affect the schedule - please reply to that. If not and the answer to the above question is no, edit the schedules to use AceCS2 instead.
XG(6.13.16): OK.
RC(6.12.16) Not clear to me what this means:
Ø The conditions selected from AU experiments, down the line, therefore, the current data can be used for comparison of the performance of Old HPLC+ New column vs. New HPLC + old column.
XG(6.13.16): Currently Alok is working with new HPLC attached with old column. XG is working with old HPLC attached with new column. So far, there is no crosstalk between these two setup. In other word, we do not compare the performance for those two machines. However, since the experimental conditions were selected from AU’s condition, we can make comparison when AU run those experiments down the line. This is just for our own record.
RC(6.13.16): Do you think use of AceCS2 (instead of p53) would allow us to explore a wider range of conditions/concentrations in initial rate studies, with more accurate quantification?
Having AceCS2, p53 and MnSOD K122 endpoint data on SIRT3/honokiol, with initial rate data on one of these substrates, will be useful for surveying the substrate-dependence of honokiol effects on SIRT3, if any.
XG(6.13.16): Two measures on HPLC Quantifiability for current two peptide substrates. First is their deacetylation efficiency by in-house SIRT3; and second is peak area. Based on results from ” Comparison of deacetylation efficiency between AceCS2 and p53 by in-house SIRT3”, under same conditions ([peptide substrate], [NAD+], [in house enzyme], incubation time), AceCS2 has more deacetylation efficiency by in-house SIRT3 than p53. But p53 peptide provides bigger peak area. Since at lower end (50uM peptide + 100uM NAD), both peptide substrates provide quantifiable results with readable peaks, I would say that they both are good for initial rate study.
Also, interestingly, AceCS2 has lower molecular weight than p53. In common, it’s retention time should be shorter than p53. However, it was not, which indicated some structure/physico-chemical properties of AceCS2 are different from p53. Therefore it is worthy to do endpoint experiments using AceCS2 for Honokiol/SIRT3 and check if Honokiol is substrate dependent activator for SIRT3.
It is a good idea to have AceCS2, p53 and MnSOD K122 endpoint data on SIRT3/Honokiol. Then we can select one of these substrates for initial rate experiments.
RC (6.7.16): Did prior substrate specificity studies, e.g. which in Nat Comm, suggest they are both equally effective as substrates in vitro?
We need to resolve this v quickly and move on with expts.
-- pending questions still remain. E.g. regarding full-length MnSOD.
Previous conversation on MnSOD:
-Regarding MnSOD protein, I saw no details in the Nat Comm paper other than a statement that antibodies specific to the acetylated and non-acetylated versions of this protein
were obtained from a group at Northwestern.
Related, do we know if it is even possible to detect and distinguish between these two versions of the full-length protein using HPLC?
AU: There are 4 deacetylation site in MnSOD (K53, K89, K68, K122) indentified by different group. There is no consensus on the primary deacetylation site by Sirt3. If all are authentic Sirt3 sites then it will be possible to see the change on HPLC provided we get really pure Ac-MnSOD full length. This will be much clean once we get rid of BSA from the buffer. I looked in the literature, some of the assay was done withought BSA. We can try this too. On side note, since peptide without fluorophore has very weak signal, we can add couple of Try at the end and acquire signal at 280 nm. Again, this will work when no other protein e.g. BSA is present and the enzyme preparation is really pure ( or should not overlap with substrate and product).
RC: 4 sites, but I thought the Nat Comm paper used anti-MnSOD (acetylated, deacetylated) antibodies -- what was meant by deacetylated...at all sites?
When you refer to literature, are you referring to HPLC studies with full length proteins substrates?
RC (5/22): Please answer the question above. I need to know whether we have enough info to replicate the conditions used with full-length MnSOD protein substrate in Nat Commun paper.
AU: 6-2-2016: Three independent group have identified 4 different sites which can be deacetylated by Sirt3. K122 was identified in Nat. Communication paper. They used K122 specific antibody to identify the site.
When I look in literature, I look for any data (assay, kinetics) which was generated using HPLC. I have not yet seen any where they used full length protein and HPLC.
RC: Confirm that the Nat Commun paper claims honokiol activation of SIRT3 deacylation of K122 in MnSOD.
XG, please work with Alok make sure the questions above regarding MnSOD and p53/other peptides we have in house are answered asap (ideally before further p53 work starts).
AU:6-9-2016: Pillai et al, used two protein show that Sirt3 deacetylate them OSCP (oligomycin-sensitivity conferring protein) and MnSOD but they used only MnSOD (K122) to test in vitro.
-- pending tasks/wiki questions will not all be repeated. you need to summarize them periodically and update.
-- proceed according to above plan without requiring day to day feedback from RC
AU:27-2016:
Next week on, I am doing Initial rate studies using p53 peptide-
1: Initial rate experiments, saturating NAD+ (2mM) with different peptide concentrations
[NAD+] 2 mM
[Peptide (p53 or FdL)]: 10, 25, 50, 100, 150, 300, 500, 600 and 1000 uM
Time: 2, 5, 10, 15, 20, 30, 45, 60, 90, 120 min
Reactions will be done with and without 10 uM Honokiol
The last two time points (90 and 120 min), and concentrations (600 and 1000 uM) may not be necessary as we might see saturation before that.
2: Initial rate experiments, saturating peptide (p53, 1 mM and it may change based on above experiment) with different concentrations of NAD.
[Peptide (p53)]: 1 mM
[NAD+]: 100, 200, 400, 500, 1000, 2000, and 3000 uM
Time: 5, 10, 20, 30, 60, 90, 120 min
Reactions will be done with and without 10 uM Honokiol
I need feedback on if I use in-house enzyme or Enzo sirt3. Although it is good to use in-house enzyme, but since Enzo has more activity which will ensure reproducibility and minimize batch to batch variation, if any.
==
AU: 5-18-2016: I will use space below to discuss MnSOD peptide synthesis and order-
AU: 5-19-2016
Some discussion points- Discussion points-Acetylation sites in MnSOD.docx
RC: What data do you have on the peak area/detectability of the peptides you currently have in-house at 280nm, given their sequences (including W,Y,F)?
You have more than just p53, do you have data on this? This would be useful to know before planning any modifications.
AU: we never planned for all peptides, should I do it? But as such readings of p53 at 280 is very less, 214 is still the best. Please note, the p53 sequence has only one F.
AU: 5-20-2016: These are the peptides I have in the lab-
P53 peptide- (Only one F in the sequence)
Pep1: NH2-KKGQSTSRHK(KAc)LMFKTEG-COOH
Pep1c: NH2-KKGQSTSRHKKLMFKTEG-COOH
H3 peptide- (No W, F or Y in the sequence)
Pep2: NH2-ARTKQTARKSTGG(KAc)APRKQLC-COOH
Pep2c: NH2-ARTKQTARKSTGGKAPRKQLC-COOH
K642acetylated human acetyl-coenzyme A synthetase 2 (AceCS2) (No W, F, or Y in the sequence)
Pep3: NH2-TRSG(KAc)VMRRLLR-COOH
Pep3c: NH2-TRSGKVMRRLLR-COO
RC: I would like to get an idea of how the possible non-native modifications will affect absorbance/peak area.
If we expect to 280 peak area to be much less even with modifications, that would make it less relevant to make/consider the modifications.
If this is not clear from the literature (see below), let me know so we can decide whether to run any other peptides prior to ordering new ones.
We will get 4 peptides one for each site, with modifications should they be useful.
We didn't explicitly discuss whether modification of the sequence (by adding such residues to increase absorbance) will alter the Km.
Although this may be unlikely, we should consider it and convince ourselves that the longer peptides will have similar affinities to shorter
irrespective of whether they use a native or non-native sequence. I would seem likely that non-native extensions of longer peptides
would have less effect (if any) on Km compared to non-native shorter peptides.
If there was any discussion regarding whether the lit reported peptides were all native sequences, let me know.
AU: I saw one report where they added 2 Ws at c-terminus of the peptide. I am not sure if they compared but this was non-native addition otherwise no need to mention.
RC: Please provide a few more details.
AU: No details except they added 2 Ws at the end for quantification purposes at 280 nm. The sequences is alpha tublin residues 36–44, H-PSDK(Acetyl)TIGGWW-NH2
RC: I didn't receive the requested info on peak area at 280 in our p53 sequence or any other. See the several questions asked about this above.
Are you able to provide any info on relative peak areas of 280 vs 214?
AU: 5-23-2016:
At 280 nm, there is no p53 either substrate or product peak which is understandable because there is no Tryptophan in the sequence.
RC: So you have no data at all on the absorbance due to these residues at 280 nm and the assumption that two tryptophans will lead to a significant peak that improves
detection is based on the fact that two were added in the past.
AU:5-24-2016: I collected the data at 280 nm but there is no identifiable peak so there are no numbers corresponding to the product and substrate peaks. Certainly, having extra Ws at the end will enhance the detectibility at 280 nm, but will it be better than 217 nm reading, I do not know. We have to scan the peptide on two different wavelengths and compare.
If we can't much more info on the questions above, we may consider ordering a couple of native 18-mer sequences (these could be ordered first)
and a couple that have non-native extensions.
AU: 5-20-2016: The sequences of 18 mer MnSOD peptide (without any modification)-
MnSOD peptides:
K53: QIMQLHHSK(Ac)HHAAYVNNL
c-K53: QIMQLHHSKHHAAYVNNL
K68: VNNLNVTEEK(Ac)YQEALAKG
c-K68: VNNLNVTEEKYQEALAKG
K89: AQIALQPALK(Ac)FNGGGHIN
c-K89: AQIALQPALKFNGGGHIN
K122: KGELLEAIK(Ac)RDFGSFDKF
c-K122: KGELLEAIKRDFGSFDKF
We can synthesise K122 peptide with 2 extra W for comparision purposes, as there are 2Fs already there, this will make more sensitive, i believe.
RC: You can go ahead and order the 4 18mer native sequences.
Let me know what the anticipated lead time is so we can decide whether to proceed with a full series of p53 initial rate expts in the meantime or not.
The nonnative 4th will be ordered after the answers above are provided.
AU: 5-23-2016: I will send these peptides for quote and ask them the turn around time. Should I synthesize control peptides too (without acetylation)? If so, this will be total 8 peptides.
RC: Yes and post the quotes.
AU: 5-24-2016: MnSOD peptide quotes, 10 mg and 25 mg. The quote includes two peptides with extra Ws so that if we need in future, we have it ready. Quote_105835.pdf Quote_105841.pdf
AU: Update: Turnaround time is 3-4 weeks. Please let me know if I go ahead and place order. Should I place 25 mg or 10 mg order?
==
AU: 5-13-2016: Proposed initial rate reactions-
1: Verify the AU18 results with p53 peptide-
NAD+ 500 uM
p53 Peptide (pep1) 250 uM
8-10U, 30 and 60 min, 37 degreeC
With 10 uM Honokiol
2: Initial rate experiments, saturating NAD+ with 10uM Honokiol/HPLC
[NAD+] 3 or 5 mM
[Peptide (p53 or FdL)]: 10, 20, 50, 100, 500, and 1000 uM
Time: 0, 5, 10, 20, 30, 60 min
Reactions will be done with and without 10 uM Honokiol
Total 84 reactions
3: Initial rate experiments, saturating peptide (p53 or FdL)+ with 10uM Honokiol/HPLC
[Peptide (p53 or FdL)]: 1 mM
[NAD+]: 25, 50, 100, 200, 400, 1000, 2000, and 3000 uM
Time: 0, 5, 10, 20, 30, 60 min
Reactions will be done with and without 10 uM Honokiol
Total 96 reactions
RC: This doesn't address issue that you can't move on with initial rate studies until you report whether you have confirmed Guan's honokiol endpoint results with FdL peptide with HPLC
or run more extensive endpoint expts with p53 peptide and honokiol to show whether there is any activation.
AU:5-18-2016- Please see table below for XG's result comparision, and for p53 end point, I updated experiment AU33, and AU34.
AU: 5-16-2016: The above proposed experiments were based on your suggestions. #1 is to see if there is any conflicting results with p53 peptide as comp[ared to FdL. I did the reaction with 100 uM NAD and 10, 100 uM FdL peptide and did not see any activation with 10 uM Honokiol (Experiment AU27). Did she repeat her honokiol experiment to see if results are reproducible (one peptide concentration gives activation and another gives inhibition, another gives no change?). With this trend, I am unbale to draw a conclusion. If you want me to repeat all her Hoonokiol experiments, I can do that too.
RC: As I mentioned you need to communicate with each other directly on the consistency of your results and who has done what experiments, when. I agree that the FdL results may be suspect given the trend.
As noted below, if there is no consistency, then after verifying feasibility of endpoint detection of products with HPLC, we may move on with saturating peptide p53/HPLC initial rate studies, and we may wait
for MnSOD peptide before doing more HPLC/honokiol experiments including initial rate experiments in presence of honokiol. It is important to order the substrates/reagents for MnSOD experiments asap (see below).
RC (5/22): When we get to honokiol/MnSOD:
We will start with endpoint studies at 10uM honokiol and one or two higher concentrations (similar series of conditions to what you previously did with FdL peptide).
Then we will do MM kinetics at 10uM and one higher concentration, thus providing the required data for a mixed inhibition fitting for honokiol
You didn't provide a summary of these issues in any ppt.
No one replied to my long posting regarding the revised schedule for all expts. And your proposed schedule above does not address this.
AU: I am searching back if I have missed something and update accordingly.
If you find conflicting results with respect to FdL/honokiol in endpoint expts, we will decide whether to proceed with saturating peptide p53/HPLC (no modulator) as a priority to demonstrate its feasibility
and since these results will be useful regardless. In that case, conditions for honokiol experiments may be determined thereafter (including decision of whether to wait for MnSOD peptides, which may
be used for subsequent initial rate studies depending on when they arrive).
I will advise on whether any DHP initial rate studies will be done after we make progress on i) feasibility of endpoint detection of products with HPLC; ii) unlabeled initial rate studies with HPLC and iii) honokiol.
RC: Waiting on i), see also below.
AU: 5-18-2016: I did the reaction using p53 peptide (250 uM), NAD+ (500 uM) with 1 uL Sirt3 enzyme on three different time points; 10, 30, and 60 min, to see 1) feasibility of endpoint detection of products with HPLC, 2) if I can use a non BSA buffer to carry out the reactions. I used HDAC buffer for the reactions. It seems that BSA can be eliminated from the assay buffer (not much difference in the product formation as compared to the buffer containing BSA, and product can be detected with above substrate concentrations (I will update 30 min time point tomorrow or any other column in these xls sheet). I used enzo enzyme because of high activity and wanted to test it quick and get clear answers.
Comparison of assay buffer and HDAC buffer (Experiment AU33)-
AU33-p53-pep1-AssayBuffer-HDAC.xlsx
RC:
AU33 updated:
| AU33-Enzo Sirt3 with p53 peptide (pep1) |
||||||
| NAD+ |
500 uM |
|||||
| Pep1 |
250 uM |
|||||
| Sirt3 |
~9U |
|||||
| Time |
60, 120 min |
|||||
| In Assay Buffer (with BSA) |
In HDAC buffer (no BSA) |
|||||
| Rxn1-Blank (120 min) |
Rxn2 (60 min) |
Rxn3 (120 min) |
Rxn4-Blank (120 min) |
Rxn5 (60 min) |
Rxn6 (120 min) |
|
| Product Rt (min) |
11.079 |
11.117 |
11.08 |
11.081 |
||
| Substrate Rt (min) |
11.639 |
11.677 |
11.598 |
11.615 |
11.628 |
|
| Product Area |
1314 |
1893 |
0 |
1292 |
1841 |
|
| Substrate Area |
7562 |
6877 |
9055 |
8009 |
7189 |
|
| Total Area |
8876 |
8770 |
9055 |
9301 |
9030 |
|
| % Product |
14.80396575 |
21.58494869 |
0 |
13.89097946 |
20.3875969 |
|
| pmoles |
1480.396575 |
2158.494869 |
0 |
1389.097946 |
2038.75969 |
|
AU34 updated:
| AU34-Sirt3-HDAC buffer |
||||
| NAD+ |
500 uM |
|||
| Pep1 |
250 uM |
|||
| Sirt3 |
11U (1uL, Enzo) |
|||
| Time |
10, 30, 60 minutes |
|||
| Rxn time 10 min |
Rxn time 30 min |
Rxn time 60 min |
||
| Rxn-1% DMSO |
Rxn-1% DMSO |
Rxn-1% DMSO |
Rxn-No DMSO |
|
| Product Rt (min) |
11.135 |
11.073 |
11.115 |
11.11 |
| Substrate Rt (min) |
11.642 |
11.602 |
11.654 |
11.658 |
| Product Area |
656 |
1205 |
1906 |
1863.4 |
| Substrate Area |
8359 |
7636 |
7237 |
6940 |
| Total Area |
9015 |
8841 |
9143 |
8803.4 |
| % Product |
7.276760954 |
13.6296799 |
20.84654927 |
21.16682191 |
| pmoles |
727.6760954 |
1362.96799 |
2084.654927 |
2116.682191 |
Assay with HDAC buffer (Experiment AU34)-
AU34-Sirt3-HDAC.xlsx
AU: 5-10-2016: Non-acetylated p53 concentration and peak area relationship- Experiment AU32- AU32-non-acetylated p53 detectibility.xlsx
I ran these samples in Assay buffer as the products will be quantified in the same buffer in actual reaction. It seems pretty linear. I will try to quantify lower than 200 pmoles e.g. 25, 50 and 100 pmoles to see if I can go further lower. It seems that even the new column started saturating with BSA which is evident by some deformed peak shape. We have to find a way to remove BSA before we load samples on the column or try different buffer without BSA or with minimum amount of BSA.
RC: You haven't done the analysis of quantities produced by the enzymes we are using in endpoint and initial rate studies (assuming similar activity to FdL peptide), which is still pending.
Without this and some commentary on how 200 pmol compares to the pmol of product expected to be formed in such reactions, I can't comment.
Also, there has been no comparison to the previously acquired data on old HPLC.
AU: 5-13-2016: Today I will be doing 10, 25, 50, and 100 pmole p53 peptide analysis on new HPLC which will complement Experiment AU32. The lowest amount of FdL peptide product I see (and can be effectively quantified in one of my experiments) is around 15-20 pmoles. I will try to get down to these numbers and see if I can quantify p53 too. In my earlier experiments, with old HPLC, I did run couple of samples, table below-
| Pep1 (Ac-p53) |
||
| 400 pmoles |
800 pmoles |
|
| Rt (min) |
15.55 |
15.433 |
| Area |
270309 |
663813 |
| Pep1c (non-Ac-p53) |
||
| 200 pmoles |
400 pmoles |
|
| Rt (min) |
14.567 |
14.767 |
| Area |
139277 |
339623 |
Au: 5-13-2016: adding 25, 50, and 100 uM p53 peptide to get area, and for comparision purposes, I ran FdL peptide with 50, and 100 pmoles.
I would say, the detection limit for p53 is between 100-200 pmoles. Stringently, I choose 200 because I can see a bump as peak for 200, but not for 100 (mostly tailing). Since the software is good so it will give me some numbers as area. In case of FdL peptide, even at 50 pmoles, the peak is really clear and well shaped.
| p53 pmoles |
Area |
||||
| 25 |
22 |
FdL pep Conc |
Area |
Rt (min) |
|
| 50 |
44 |
50 |
103 |
12.315 |
|
| 100 |
44 |
100 |
204 |
12.313 |
|
| 200 |
123 |
||||
| 400 |
326 |
||||
| 500 |
460 |
||||
| 600 |
522 |
||||
| 800 |
702 |
||||
| 4000 |
3800 |
RC: This is not very informative, esp as far as initial rate studies are concerned. What is the answer to my question regarding whether initial rate experiments will be possible with p53? New hplc? Old hplc?
Why/why not? What is the basis for conclusion that above initial rate experiments will be detectable on new hplc?
Or do you need more active enzyme?
AU: 5-18-2016: I think the initial rate using p53 is possible (please see AU33, and AU34 experiments). Also, it would be better if we have high activity enzyme.
RC: I need more elaboration on this as well the detailed comparison of what peptide concentrations/amounts of p53-like peptides will be detectable by
a) old HPLC; b) new HPLC, with reference to whatever data and analysis you previously did with old HPLC. Based on this analysis, I would like to see a detailed commentary
on what types of endpoint experiments will be feasible with p53 peptide on both old and new HPLC, as well as a detailed commentary on what types of initial rate experiments
will be feasible on old and new HPLC.
AU: 5-23-2016:
Based on the table posted above, Old HPLC, p53 peptide, detection linit is 200 pmoles or more whereas for new HPLC it is more than 100 pmoles. As far as endpoint assay/initial rate experiments feasibility is concen, as long as I get product formation in the range (100 or more pmoles for new HPLC, 200 or more for old HPLC) the any experiment is possible. e.g. endpoint assay with 500 NAD+, 250 uM p53 peptide in 5-10 min or longer reaction time is possible. For initial rate, if we use same NAD 500 uM (or more) but change the peptide concentration to 10 uM and assume the reaction rate doesn't change (NAD 500, peptide 250 pmole, product % = 7.2%), the total product will be 500 pmoles x 7.2% = 36 pmoles which can not be detected on either HPLC. Since readout is peptide product formation, the feasibility of experiment may be more when I use saturating peptide and varying NAD concentration. But the situation is different for initial rate where one of the substrate is saturating (>2mM NAD or >1mM peptide), the behaviour of enzyme may unpredictable ( I do not know how enzyme will react in different experimental conditions.
RC: You need to immediately provide a comparison to literature HPLC initial rate studies on sirtuins, indicating that if other sirtuins were used how they were able to quantify products under the conditions used (including whether lower substrate concentrations were detectable) based on what you know about the enzyme (sirtuin isoform) activity differences. Your statement that is is unclear whether we can run initial rate studies under various conditions is concerning, as I have stated several times before, though perhaps this is attributable to your assumption of very low peptide concentrations.
Ideally, you would have explored this issue with tech support prior to equipment purchase.
AU: 5-24-2016: I am still looking into the literature to find out initial rates done using HPLC. Meanwhile I can say that the initial rate studies using saturating NAD (2mM) and p53 peptide is doable using HPLC. I used Enzo Sirt3 (11U/rxn) to do the reaction and I could quantify the products after 5 and 10 min time at two different peptide conc. 10 and 30 uM. 10 uM did not give me linear relationship between 5 and 10 min. I suspect that the enzyme activity may be too high (speculation). We can start with 20 uM peptide and start the reaction starting from 2 minutes. Please see data for experiment AU35: AU35-Test initial rate.xlsx.
"e.g. endpoint assay with 500 NAD+, 250 uM p53 peptide in 5-10 min or longer reaction time is possible": how much product are assuming will be formed in this reaction?
"change the peptide concentration to 10 uM and assume the reaction rate doesn't change (NAD 500, peptide 250 pmole, product % = 7.2%), the total product will be 500 pmoles x 7.2% = 36 pmoles": please discuss this with XG and verify.
Also, if peptide concentration decreases, reaction rate will change.
It will be important to know for both endpoint and initial rate, which conditions (concentrations of reagents, and also time in case of endpoint) we expect will be detectable.
You will collectively be responsible for ensuring that we can detect initial rate products with HPLC for all the conditions required in the priority experiments in our plan that has been specified.
AU: 5-24-2016: Yes.
RC: Note that Guan should have data on relative amounts of products formed at 5-10 mins under all different combinations of concentrations used in our previous initial rate experiments (saturating peptide).
AU: 5-26-2016: I will ask her to share her data of initial rate, but did she do the assay using HPLC? or it was FdL based assay. If FdL based assay the the amount of product formed will be different. Also, I can confirm that the initial rate studies are possible using HPLC method.
RC: As you know we did not do initial rates with Hplc in the past. FdL substrate does not only apply to FdL assay. This comment was to facilitate analysis if helpful.
Please indicate today the status of this task.
AU: 5-27-2016: XG's initial rate data: Enzo SIRT3-Control-XG-data.xlsx
Table below is extracted from above file.
| Read out unit: uM |
|||||
| [NAD], uM |
|||||
| Time, min |
100 |
375 |
750 |
1500 |
3000 |
| 0 |
0.000 |
0.000 |
0.000 |
0.000 |
0.000 |
| 10 |
0.103 |
0.411 |
0.565 |
0.640 |
0.809 |
| 20 |
0.416 |
1.104 |
1.428 |
1.974 |
2.113 |
| 30 |
0.557 |
1.702 |
3.092 |
3.791 |
3.899 |
| 60 |
1.118 |
3.316 |
5.604 |
6.050 |
7.177 |
| 120 |
2.304 |
6.147 |
7.781 |
9.284 |
10.033 |
| 180 |
3.060 |
8.034 |
11.158 |
11.847 |
12.449 |
| 240 |
3.511 |
9.491 |
12.061 |
12.463 |
13.371 |
As you know these are relevant to planning of next steps.
RC (5/22): I still do not have an answer regarding whether we will be able to do initial rate studies with SIRT3 on the new HPLC.
More analysis is possible without actually doing the experiments.
I also do not have answers regarding comparison of endpoint detection limits of old and new HPLCs, and the conditions under which reaction products will be detectable given the information on the minimal peptide concentrations that are detectable.
AU: 5-23-2016: Please see above and two tables to compare the limit of detection. The only thing I will add is that with old HPLC, when I ran 100 pmoles p53, it was not very clean and was similar to blank reading that is why I say by old HPLC, the product more than 200 pmoles will be reliable (at-least, when I did that run). Nonetheless, it is possible to do initial rate with Sirt3/p53 peptide on new HPLC.
RC: I was referring to reaction products under enzymatic assay conditions. The critical question is how much product is formed (and whether it will be detectable) under those conditions.
After providing this as requested, you can start by proposing just one initial rate experiment (one set of concentrations) with p53 peptide and indicate when you will be able to analyze and report the results.
AU: 5-23-2016: I will do initial rate with varying NAD and saturating (assuming 1 mM p53 will be saturating) peptide. Should I use in-house enzyme? Please suggest.
RC: Only in-house can be used for publication. If you think commercial will allow for better sensitivity, indicate why and what type of improvement is expected in detail.
AU: 5-24-2016: Theoratically It should not matter. Since I want to use minimum protein, as BSA was eliminated and works well, using equivalent activity in-house (total 10U; ~1U/uL) will have more protein as compared to enzo (~11U/uL). That is why I asked this question.
RC (5/9):
--I am receiving piecemeal answers to many of my questions. At the end of last week I had asked for clarity on when answers to all the questions will be provided
since they are critical for planning.
--Last Sun, I had posted many such questions and tasks, including:
-Guan was supposed to repeat the honokiol experiments with FdL, verifying whether or not she saw activation under the same conditions as last time.
XG(5.9.16): Before to repeat the honokiol experiments with FdL, the effect of 1% DMSO to SIRT3 need to be validated. As mentioned below, Delta AFU increased by addition of up to 35%DMSO. However, the decrease of Delta AFU was detected in the presence of 1% DMSO. That's why the repeat of DMSO experiments are important.
RC: See below. I mentioned a couple of times it could be repeated with HPLC. I don't believe you replied to that. Note my comments regarding possible attribution of decrease in Delta AFU to increase in peptide Km. Note that the inhibitory effect of 1% DMSO decreased with increasing [peptide], which may be consistent with an increase in peptide Km.
Regarding higher % DMSO, not only did Delta AFU increase, but AFU at t=0 increased. I asked about autofluorescence effect in this regard, but there was no suitable answer.
-Questions were posted regarding autofluorescence effects of DMSO, which could render the results inconclusive/false positives. These were not all answered to satisfaction.
Repeating those DMSO experiments with FdL was not the immediate priority. In particular it was mentioned that HPLC (e.g., old HPLC) could be used to verify or invalidate the DMSO effects
on activity. This was never planned. I had mentioned that either Guan or Sudipto could do this if Alok was busy.
-Alok was to redo some of Guan's honokiol experiments with FdL substrate and then summarize the results.
Some of this was posted but I would like to see a very brief summary indicating which experiments were repeated and whether activation was observed under the same conditions Guan observed
it, or not. Ideally, by the time some of these experiments were done, the FdL experiments would already have been repeated for verification since FdL is faster, but that was not the case.
AU: 5-17-2016: The following experiment was done for Sirt3 (1ul) and Honokiol 10 uM.
| 100 uM NAD+, 100 uM Peptide |
100 uM NAD+, 50 uM Peptide |
100 uM NAD+, 10 uM Peptide |
|||||||
| Rxn10-No DMSO |
Rxn11-1%DMSO |
Rxn12-10uM HNK |
Rxn6-No DMSO |
Rxn7-1%DMSO |
Rxn8-10uM HNK |
Rxn2-No DMSO |
Rxn3-1%DMSO |
Rxn4-10uM HNK |
|
| Product Rt (min) |
10.602 |
10.565 |
10.574 |
10.638 |
10.648 |
10.646 |
10.504 |
10.506 |
10.664 |
| Substrate Rt (min) |
12.256 |
12.258 |
12.262 |
12.312 |
12.303 |
12.303 |
12.397 |
12.241 |
12.343 |
| Product Area |
76 |
77 |
80 |
74 |
56 |
64 |
28 |
29 |
21 |
| Substrate Area |
7701 |
7714 |
7538 |
3862 |
3836 |
3801 |
749 |
748 |
729 |
| Total Area |
7777 |
7791 |
7618 |
3936 |
3892 |
3865 |
777 |
777 |
750 |
| % Product |
0.977240581 |
0.988319856 |
1.050144395 |
1.880081301 |
1.438848921 |
1.655886158 |
3.603603604 |
3.732303732 |
2.8 |
| pmoles |
39.08962325 |
39.53279425 |
42.00577579 |
37.60162602 |
28.77697842 |
33.11772316 |
14.41441441 |
14.92921493 |
11.2 |
AU: 5-18-2016: I did three different concentrations of FdL peptide and do not see any change in activity with 10 uM honokiol. These concentration are listed in the table above. And all three was also done by XG.
-Guan was to move on with initial rate experiments using honokiol/FdL if Alok confirmed the trends she saw with HPLC.
-Alok was to move on with p53 studies in the meantime. This is because we need to verify p53 quantifiability before moving on with MnSOD peptide/protein.
This was to start with endpoint studies and an indication/analysis regarding quantifiability of endpoint and initial rate products (in that order) with old and new HPLC.
This was to include e.g. analysis with old HPLC p53 data in hand on what concentrations can be quantified. This was not completed.
--Since Alok said he was too busy to do all the necessary quantifiability/standards analysis on HPLC alone, Sudipto was asked to contribute to some of this analysis in the meantime as needed.
A schedule for that is also requested.
-Further advice on schedule of Sudipto could not be provided since I had also asked for clarification on how much in house enzyme would be required for the proposed experiments and how much
of Sudipto's time this would take. Then we could assess if he could assist with HPLC quantifiability analysis in parallel.
All proposed initial rate experiments were to be done with in-house enzyme.
-The schedule now needs to be revised per the various incomplete tasks above. However, note that I cannot possibly advise on every detail of the order of these experiments and how that order should
be updated every couple of days based on incomplete experiments altering the order.
-Since Guan has not yet repeated honokiol/FdL experiments, the only circumstance under which Alok should start initial rate experiments with p53 is if he saw some confirmation of Guan's honokiol/FdL results last week.
If not, the next HPLC work should focus on quantifiability of products (both endpoint and initial rate) and p53 quantification analysis on new HPLC. Detailed analysis should be provided indicating what we can detect on each HPLC
for p53 substrate.
DMSO study on HPLC should also be done by someone.
Alok would then subsequently proceed with honokiol/HPLC endpoint experiments with p53 substrate. This would need to include more than just one set of concentrations.
We would then proceed with initial rate studies thereafter based on the endpoint results analysis.
-Based on whether Alok was able to confirm Guan's results with honokiol/FdL, we will consider whether Guan should move on with honokiol/FdL initial rate studies for do more work on assay development/standardization and contribute
to HPLC assays.
AU: 5-9-2016
Sudipto will let me know today how much enzyme (amount, concentration and the activity) he has and then I will start with p53 peptide and post a schedule soon after.
XG is doing initial rate (with Honokiol) with FdL peptide, as per her schedule whereas I will be doing with p53 peptide.
Question: Do I have to repeat all my Honokiol experiments with p53 peptide with in-house Sirt3 enzyme, before I proceed to initial rate studies? Thank you.(I assume only experiment AU18 where I used 500 uM NAD, 250 uM FdL peptide and the reaction was done for 60 min. The only change will be the peptide).
My next step will be to do following experiments, as suggested by Raj-
"If so, the next steps with HPLC may be to 1) verify the above results with p53 peptide; 2) run two types of initial rate experiments with 10uM honokiol/HPLC (and ideally p53 peptide
if possible): a) saturating NAD+; b) saturating peptide, using Sudipto's enzyme.
(see XG's lab page for more details).
MM fittings for these should be provided.
Indicate how much in house enzyme will be needed and if you have any concerns about running out or that sudipto will need to work full time on just purification to provide enough, indicate that immediately
Hplc quantifiability of initial rate products using sudiptos enzyme should be validated as a priority. If there are any concerns indicate them immediately with thorough explanation.
The deadline for these expts is tentatively set to 1 month. Provide the schedule for all the above accordingly.
It will be finalized after receiving info on time required for a single set of initial rate expts (multiple nad or peptide concentrations) on Hplc.
Two hplcs will have to be run in parallel or one on a staggered schedule"
===
AU: 5-4-2016: Experiment AU31-Sirt3-DHP1c:
AU31-Sirt3-DHP1c.xlsx
AU: 5-4-2016: Is it possible to do quick docking of Honokiol and other compounds to Sirt3? Thank you.
AU: 5-4-2016: Experiment AU23-Sirt3 Honokiol-
Au23-Sirt3-Honokiol-Final.pptx
The above reaction was done with 100 uM NAD+, 100 uM FdL peptide at 37 degreeC for 60 min using 5 uL Sirt 3/Reaction (40U as per Enzo's specification). The activation was not detected, we have to re-work on the assay protocol to confirm if Honokiol truely activates the enzyme or not. This will require a lot of active enzyme and couple of weeks time to find a balance between concentration and reaction time. This will also enable us to proceed with the initial rate studies.
Today I am planning to do the Sirt3-DHP1c using 100 uM NAD and 100 uM peptide at 37degree for 60 min reaction using 5 uL Sirt3. I will try to post the conclusion of the the experiment, and if possible I will post the data.
RC: I mentioned on Guan's page need for Hplc validation of the various FdL honokiol experiments. Which experiments are you able validate? Only the one you did so far? We need to know whether her honokiol results are validated before she moves on with initial rate studies on it, as noted on her page. Comment on why new Hplc does not seem to have better sensitivity than old for these assays (assuming you are observing this). This is why I asked you several times to establish the detection limits. What is your issue with detecting product at 10uM peptide? If you need to do that analysis this week before Sudipto has time to look into it next week please do so and report.
AU: I have done several experiments with different substrate combinations, with different amount of enzyme. Since my experiments and output is time taking, I just posted one. Please see another here. As far as comparision of FdL and HPLC is concern, she used minimum 5U/reaction and got something. I used minimum 8U/reaction and the product is really low but I can detect it with new HPLC which was not seen with old HPLC (background was too noisy). This also suggest that new HPLC has improved the quality of the data which is related to the sensitivity is concerned. There is no issue with 10 uM peptide in this HPLC, it is just I did not upload it, I will upload as soon as I finish analysing it. As far as experimental validation of XG's data is concern, I have matching reactions but I am analysing the data right now and I will post it here (Update AU27-Sirt3-Honokiol.xlsx DATA with 1uL Sirt3: 10uM peptide-100 uM NAD). I looked her data, she has to confirm what she got, gets everytime she does the experiment. Please see e.g (extracted from the data she sent to me).XG-Honokiol-data.xlsx
This reaction was done two different peptide concentrations, 10, 100 uM and NAD was fixed with 2.5 uL of enzyme. AU28-Sirt3-Honokiol.xlsx
Related, "rework the protocol" for sirt3/Hplc is not yet clear. Same about proceeding w/ initial rate studies. You know the activity and you also saw how people did initial rate expts w sirtuins in lit.
We need verification of feasibility of initial rate studies shortly.
AU: "re-work" means e.g. I have not done the saturating NAD (2mM) and titrate peptide concentration on different time points and vice versa. I am confidant that the initial rate for Sirt3 can be done with FdL peptide. We may not be able to get some initial time points e.g. 5 or so min but we can get good initial rate data. Since Sudipto is going to do peak area conc relationship with non FdL peptide, we will get some idea from there.
There are still pending questions on MnSOD and well as several other questions posted this week on wiki.
AU: 5-5-2016: As noted earlier that the changing peptide length from 5 aa tp 8 amino acids does not affect Kcat/Km values. Also, changing peptide length from 18 to 12 does not affect Kd values significantly.
A survey of several paper suggest that most of them used 11 aa length peptide (Bheda et al., 2015: UPDATE: List obi85_lin_suptable4.xlsxf Sirt3 substrate proteins, I looked most papers cited, experiments seems to be done in vivo, I will keep searching and update here if I find any in vitro study were done. ). It is hard to say "why" they choose a particular peptide but I would conjecture they must have tried few and used what worked for them.
RC: I would like to see a list of perhaps 10 of the peptide sequences used in previous papers posted to wiki, so we can use that to facilitate choice of sequence for MnSOD peptide.
AU: I looked at the literature, and could not find at-least 10 peptide used in in vitro studies. Most of the people used one of p53, H3 (and H4, for H4 there was a mention in review article but the sequence and reference was not given), or AceCS1/2. I will keep searching and if I come across with any new peptide, I will upload it here.
The sequences are-
| ARTKQTARKSTGG(acK)APRKQLC |
Landry et al., 2000 |
| Ac-KQTARKacSTGGKAPR-WW-NH2 |
Feldman et al., 2015 |
| H2N-KRLPKTRSG-AcK-VMRRLLRKII-COOH |
Hirsch et al., 2010 |
| TRSG-AcK-VMRRL |
Nguyen et al., 2013 |
RC:
-It is important to refer to the database of ~6000 peptides that Steegborn and others have studied. I believe we discussed this paper earlier and it is cited here.
AU: 5-16-2016: They used 13 mer peptide in the library (Rauh et al., 2013).
-It is true that for SIRT3 the datasets suggests 5 residues flanking either side of acetyl-Lys residue, for a total of 11, is common and may be sufficient for specificity.
-But what are the disadvantages if any of using longer ones? Some of the 4 you list above have more.
AU: 5-16-2016: I don't know what are the disadvantages would be if longer peptide is used. But as mentioned earlier, Garske and Denu (2006) say that changing the peptide length from 5 amino acid to -8 amino acids does not affect Kcat/Km values whereas changing peptide length from 18 to 12 does not affect Kd values significantly as described by Cosgrove et al., 2006 .
-You mentioned most papers cited in the review did in vivo studies, but there is a section on in vitro studies on peptides. Where did you get this xls table from in that regard?
AU: 5-16-2016: The table is available as supplimentry material in the review article.
RC: The relevant question is, does this table pertain to the section on in vitro studies on peptides?
-We should not use diacetylated peptide at this time.
AU: 5-16-2016: OK
-We should list here some peptide sequences between 11 residues and the longest you see in the reported literature, with equal numbers of residues flanking either side of the acetylated Lys.
-We will probably order several of these. We will then check MM plots at saturating NAD to determine whether peptide Km is roughly the same or different for these peptides.
AU: 5-16-2016: Please see above, changing peptide length does not change kinetic parameter or Kd significantly.
RC: Ok, but we may still order at least a couple of different lengths if we identify a single site. If we do not know which site is the relevant one, we can order four peptides on the somewhat longer site (to be safe) for the four sites,
unless you see a potential issue with length.
Please make preparations to order shortly (see also below regarding acetylation site).
-Regarding MnSOD protein, I saw no details in the Nat Comm paper other than a statement that antibodies specific to the acetylated and non-acetylated versions of this protein
were obtained from a group at Northwestern.
Related, do we know if it is even possible to detect and distinguish between these two versions of the full-length protein using HPLC?
AU: There are 4 deacetylation site in MnSOD (K53, K89, K68, K122) indentified by different group. There is no consensus on the primary deacetylation site by Sirt3. If all are authentic Sirt3 sites then it will be possible to see the change on HPLC provided we get really pure Ac-MnSOD full length. This will be much clean once we get rid of BSA from the buffer. I looked in the literature, some of the assay was done withought BSA. We can try this too. On side note, since peptide without fluorophore has very weak signal, we can add couple of Try at the end and acquire signal at 280 nm. Again, this will work when no other protein e.g. BSA is present and the enzyme preparation is really pure ( or should not overlap with substrate and product).
RC: 4 sites, but I thought the Nat Comm paper used anti-MnSOD (acetylated, deacetylated) antibodies -- what was meant by deacetylated...at all sites?
When you refer to literature, are you referring to HPLC studies with full length proteins substrates?
RC (5/22): Please answer the question above. I need to know whether we have enough info to replicate the conditions used with full-length MnSOD protein substrate in Nat Commun paper.
=
RC: We do not need to restrict to only SIRT3 substrates. Do you mean there are only 4 sirtuin substrates listed in the literature for all sirtuins?
AU: The one I listed (4 substrates) are for Sirt3 and the experiment was done in vitro.
Please indicate which peptides were used with which sirtuins and highlight the ones used for SIRT3 in vitro.
Was there any comparison of activity on peptide and protein substrates? It would be useful to see at least one example of that since the only data in lit for MnSOD is with the full-length protein.
I see you are referring to the recent paper surveying the sirtuin substrates when you mention most experiments were done in vivo, but certainly many initial rate studies have been reported, no?
I will review both the FdL and non-FdL substrates for ordering after receiving an update on this.
Note also that verification of p53 product detectability is important before moving on to other non-FdL substrates.
AU: I will try to finish the p53 product detectibility limit by the end of the day tomorrow (Updated 5-10-2016), and I will update ansrwers to other questions as I gather more information.
==
AU:5-10-2016-I looked at the articles mentioned in Bheda et al., and none of them did kinetics except one (Hallows et al 2011) who determined the Km peptide but failed to mention the sequence used in the experiment. None of the study compared peptide and protein in their studies. All of them used western blot for their read out. Following may be helpful-
Vassilopoulos et al., 2014, ANTIOXIDANTS & REDOX SIGNALING Volume 21, Number 4, 2014
(Used peptide as a substrate in vitro experiment but detected by mass spec)
The specificity of this lysine deacetylation was determined using recombinant human SIRT3 and a synthetic peptide ( JPT Peptide Technologies) corresponding to a 20- amino-acid Oligomycin sensitivity conferring protein (OSCP) sequence containing lysine 139. This peptide (sequence EEATLSELKTVL(AcK)SFLSQGQ) was incubated with recombinant SIRT3 (BioMol, Inc.) and NAD + , and the reaction was monitored by MALDI-TOF measurement of the peptide mass (Fig. 3c). After incubation of the acetylated synthetic peptide with recombinant SIRT3, all of the peptide was reduced in mass by 42 m/z. A control reaction with nicotinamide (an inhibitor of sirtuins) caused no shift in the signal, nor did a control with NAD + and no SIRT3 (data not shown). Similar experiments were performed on a related peptide with acetylated threonine instead of a lysine (sequence EEATLSELK(AcT)VLKSFL SQGQ), with no evidence of deacetylation (Supplementary Fig. S3A).
Hallows et al., 2011, Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction, Mol Cell. 2011 Jan 21; 41(2): 139–149.
Kinetics was done on OTC-K88 peptide (Figure 2A) with Sirt3 but no specific sequence was provided. Looking at the supplement information, a peptide can be identified (MIFEK*RSTR) which was mentioned by Bheda et al.
Inuzuka H, et al. 2012. Cell 150: 179
Used biotinylated peptide 49-88 for binding assay and deacetylation with western blot read out.
Zhang et al., 2015, PlosOne SIRT3 and SIRT5 Regulate the Enzyme Activity and Cardiolipin Binding of Very Long-Chain Acyl-CoA Dehydrogenase
“peptide identification was done by mass spec”
Both SIRT3 and SIRT5 efficiently targeted the peptide GFGGITHGPPEKKMGIK303 when doubly acetylated or succinylated at K298/K299. An examination of the VLCAD crystal structure reveals that K298 and K299 are located near the active site in close proximity to the FAD cofactor (Fig. 2A).
=
RC: Previously we discussed more than once the need to post the old honokiol results with HPLC.
To my knowledge these data were never posted. It is of course not possible to compare the FdL results with the HPLC results
if the data are not posted. If they were, please indicate where. If not, why not despite the various requests?
Au: 5-2-2016: Please see Experiment AU18 data (This experiment was done one time to see if Honokiol activates Sirt3 with 500 uM NAD and 250 uM peptide)- AU18-Sirt3-Honokiol.ppt AU18-Sirt3-Honokiol.xls
RC: Given that the schedule posted earlier indicated the specific experiments in each case with HPLC would depend on the FdL results,
please provide the detailed plan for honokiol/HPLC experiments to be done this week (with new HPLC I assume). I would like to know which of the FdL experimental
series with honokiol they will repeat. I may then have some comments in this regard.
AU: 5-3-2016: Although Sirt3 has lower activity, the fluorescent based assay will give some readout even with 100 uM NAD, 10 uM peptide. For HPLC, it is unlikely. The HPLC assay will cover some of the points and match the experimental conditions used in FdL assay. Right now I am doing reaction with 100 uM NAD, 10 or 100 uM FdL peptide. I tried to titrate the amount of enzyme and repeat it today. I see inconcistency in the results ( I think activity of these two batches of Sirt3 may be different).
We can draw a rough estimate of the concentrations Nat Comm paper used e.g.
Along with honokiol/HPLC experiments, DMSO effects on SIRT3 as identified by FdL (see the recently reported honokiol/FdL experimental data) will need to be validated with HPLC.
AU: 5-3-2016: I did not include in these reactions but I will include in next set of the reactions.
Sudipto will meanwhile be verifying that p53 peptide product can be detected and initial rate experiments can be run with this HPLC.
If so, the next steps with HPLC may be to 1) verify the above results with p53 peptide; 2) run two types of initial rate experiments with 10uM honokiol/HPLC (and ideally p53 peptide
if possible): a) saturating NAD+; b) saturating peptide, using Sudipto's enzyme.
(see XG's lab page for more details).
MM fittings for these should be provided.
Indicate how much in house enzyme will be needed and if you have any concerns about running out or that sudipto will need to work full time on just purification to provide enough, indicate that immediately
Hplc quantifiability of initial rate products using sudiptos enzyme should be validated as a priority. If there are any concerns indicate them immediately with thorough explanation.
The deadline for these expts is tentatively set to 1 month. Provide the schedule for all the above accordingly.
It will be finalized after receiving info on time required for a single set of initial rate expts (multiple nad or peptide concentrations) on Hplc.
Two hplcs will have to be run in parallel or one on a staggered schedule
RC: While doing the honokiol experiments this week you should reply regarding the MnSOD peptide-related questions below so we can move that forward
and make the relevant decisions. Analogous initial rate studies will be done with this substrate after finishing the above.
AU: 5-2-2016: I will update some of the concerns tomorrow and rest of the (schedule and amount of enzyme needed for the experiments) as I get time.
AU: 4-29-2016: Experiment AU25- Sirt1 and DHP1c
AU25-DHP1c-Sirt1.pptxAU25-DHP1c-Sirt1.xlsx
RC: These can be re-run after the honokiol experiments.
We may then need to also do the two types of Km, vmax experiments above with DHP/SIRT1 to establish their effects vis-a-vis the JMC paper 2.
This would be a mixed inhibition type of experiment (with commercial enzyme) to establish the mode of inhibition if the compounds are just inhibitors not activators.
It could be run around the IC50 of the DHP as determined by HPLC should in the event that there is no evidence of activation at the concentrations
used in JMC.
If we prefer to use in-house enzyme we may do these experiments on SIRT3 instead.
DHP1c/SIRT3: Based on the results above with DHP1c/SIRT3, the reported 50 uM is nowhere close to IC50. 50uM may however be enough for a study aimed at showing lack of activation as claimed in JMC, if desired.
In future, we would need to do significantly higher concentrations (noting solubility limit) to find IC50 for purposes of initial rate assays if desired. Could be done with either SIRT1 or 3 (required concentrations for SIRT1 may be lower).
RC (4-29):
--I assume you have finished running your standards on new Hplc. Please post that data.
Does that include # 3 below?
AU: 4-29: I am not running #3 on new HPLC, If I do that I will not be on schedule for the deadline. Even though, I may not be able to finish all proposed experiments by Wednesday next week. I would request you to consider the deadline extention, at-least another week.
--Also, provide an update on which expts are now being run. Next week I will be running Sirt3 titration reactions and then Honokiol experiments. Please consider deadline extention. Thank you.
RC: Before commenting on deadline extension, both you and XG should specify how many hours you worked per week since the time of our last meeting when the deadline was specified.
AU: 4:29: Currently, I am running DHP1c-Sirt1 reactions. I will send out data today once I finish it. It will be in the for of Excel. Since I am learning new software as I do the experiments, I do not have time to explore how to transfer chromatogram in to powerpoint.
--Given these results, can we now conclude that we can detect initial rate products as in the literature on new HPLC?
AU: 4-29: For Sirt1 yes, and for Sirt3 we have to modify the protocol to get the initial rates. UPDATE on Old HPLC: Scott will be in the lab on Monday to fix the system.
RC:What about posting the data on the standards? I didn't see a reply on that question.
I am talking about all the standards you ran so far on new HPLC.
AU: 4-29: I am not running standards, I ran blank samples to condition the column, clean up the buffers in new HPLC, then ran the blank samples again to see if retention time is stable. I ran one concentration of NAD (with and without DMSO) to see if there is any change in retention time. Then I started running the reactions on the HPLC (since yesterday, I am running Sirt1-DHP1c experiments). As such I did not run the standard (reactants alone) to get the peak area and concentration relationship. This will be done once we finish the proposed experiments.
RC: Then Sudipto will need to start this soon.
Depending on your replies, we may ask Sudipto to work on the new HPLC during hours when you are not working,
so as to accelerate progress on getting the relevant data to answer some of these questions.
AU:4-26-2016
OLD HPLC update-
Even with new column, there is some inconsistency in the chromatogram (e.g. the peaks are not resolving properly) and seems that there is back pressure building up even only one pump is working. I contacted Scott and he told me that there might be internal leak in check valve. I sent him a text message to see if he can visit us and fix it. I am still waiting for his response. Please see text messages here. Scott conversation.pptx
Since I have limited time, I moved the column to new HPLC and testing the method and see If I can resolve the peaks properly. If so, I will run rest of the samples (reaction with Sirt1/DHP1c) to get the data. If something not right, then I will try to modify the program.
AU: 4-21-2016
"
Garske and Denu (2006) say that changing peptide length from 5-8 amino acids does not .."
AU: This should read "changing peptide length from 5 amino acids to 8 amino acids does not affect Kcat/Km...." i.e. if they used peptide with 5 amino acids or 8 amino acids, the kinetic constant was similar.
I will update on
1: why a particular peptide was chosen, if I find it.
RC (4/29): Ok, please provide this info so we can move forward with this.
2: p53 peak area relationship with concentration (old HPLC)
3: p53 peak area relationship with new HPLC. I need to work on new HPLC and run the sample. Length of peptide papers bi052015l-Garske-2006.pdfbi0526332-sirt substrate affinity 2006-cosgrove.pdf
RC: Regarding detectability of unlabeled products with the HPLCs, what about with purified enzyme assuming e.g. the activity of Sudipto's latest batch (instead of Enzo)? (Comment on endpoint and initial rate.)
AU: Yes, the experiments can be tried with enzyme purified by Sudipto.
I am looking for an analysis that mentions In house enzyme (given its specific activity) and initial rate studies in the peak
area discussion you will be adding per your comments below.
AU: If I understand the comment above, the response is- I never did any initial rate studies with any enzyme, even after 2 hours of reaction with 8M urea purified enzyme, the product was too low to detect. I will upload the p53 (alone, not in a reaction) peak area v/s concentration once I get time. (please let me know if I understand the comment, thank you)
RC: Again, I asked you to indicate, given what you know about the activity of the new in house enzyme and Guan's initial rate data, whether the initial rate studies would be possible with that HPLC.
You already presented p53 data earlier. You now need to comment on its detectability given the previous calculations you did, referring to that data (#2 above).
"Previously with old Hplc you were of the opinion initial rate studies would probably not be possible"
AU: Yes and it was in reference to Sirt3 enzyme.
RC: The name of the enzyme is not as directly relevant as is the activity.
We've discussed this before. You need to extrapolate based on what you know about the enzymatic activities.
"and that in house enzyme may not be detectable even in endpoint studies."
AU: I tried one time, 2 hour reaction with in house (8M urea purified) enzyme and the peak was too small and not distinguishable from the background. Then we moved to Enzo Sirt3
RC: Again, you have activity data. You are referring to trial and error approaches. You implied the activity was too low for detection.
RC: Regarding full-length MnSOD protein, please comment on whether use of this in HPLC could lead to column saturation.
You previous made several comments on how protein may saturate the column.
AU: That was in reference to high amount of BSA (total protein) in the assay buffer (BSA is 1mg/ml in assay buffer). If we remove BSA by any method, the amount of substrate (MnSOD) will not affect drastically. Since MnSOD will be used as substrate , the amount will be low, e.g. upto 500-1000 uM or so in saturation reactions. These amount will be effective (causing saturation) in long run and much delayed as compared to having BSA.
RC: So is it more important to remove BSA if we plan to use full-length MnSOD protein as a substrate?
If so, do you have a protocol for removal?
AU: 5-5-2016: Yes, BSA should be removed regardless we use full length MnSOD or not. This will save column life. One method, I mentioned earlier to filter it out. This will require some optimization to see how much substrate/product filtered out along with the protein.
===========
Email: Wed 4/20/2016 10:56 AM
“AU: Garske and Denu (2006) say that changing peptide length from 5-8 amino acids does not affect Kcat/Km values whereas changing peptide length from 18 to 12 does not affect Kd values significantly as described by Cosgrove et al., 2006 .
“
Please elaborate on this, and put the relevant replies and pdfs on wiki. (peptide length was 5-8 amino acids?) Include basis for choice of sequence (not just length) where relevant.
Regarding detectability of unlabeled peptides, you should provide more details regarding detectability on both HPLCs on the wiki. For old HPLC, this should refer back to your previous notes on calculation of peak areas.
Priority should be completion of your deadline experiments. Schedule for that is not clear.
==========================
Tuesday, April 19, 2016 3:02 PM
-- Regarding
(1) Auto-fluorescent controls A: Enzyme, substrate, NAD+, and incubate at 37oC for desired time points. Then add the compound at the same concentration as its experimental counterpart. Then developer and read.
(2) Auto-fluorescent controls B: Enzyme, substrate, NAD+, and incubate at 37oC for desired time points. Then add developer and read. Then add the compound at the same concentration as its experimental counterpart and read.
(3) Auto-fluorescent controls C: substrate, NAD+, and the compound at the same concentration as its experimental counterpart.
C) has the following issue: it does not account for fact that AFU of DHP + deacetylated product is not necessarily equal to AFU of DHP plus AFU of deacetylated product.
This is why A,B were suggested (though as noted they too have problems).
As noted you need to look further into the possible use of synthesized deacetylated FdL peptide as a standard. Standard curves with this would need to have all the other relevant components
of the reaction mixture. Controls A,B) were suggested partly because we did not know the identity of the FdL standard and hence it could not be used in this way.
You need to consider this and provide feedback on whether it would be sufficient/rigorous in absence of controls of type A-C) above, which seem to have flaws. This includes
analysis of other sources of uncertainty/variability like developer chemistry and whether the standard could be run almost identically to experimental sample with exception of substrate.
If you are currently claiming that FdL works fine for any modulator without intrinsic fluorescence, please state that for the record.
Related, you need to look into other assays including Sirtainty and answer the pending questions regarding whether the manufacturers have fully disclosed the identities of the reagents used,
identification of the standards used and possible issues with them, and what would be suitable controls for the preferred assays with indication of advantages over FdL.
As noted this analysis should be done separately for modulators with and without intrinsic fluorescence.
Note in this regard that the 2015 Nat Comm paper on SirReal inhibitors/SIRT2 discussed a fluorescence-based assay that was apparently carried out with only in-house reagents and not kits.
Such assays that can be implemented fully in-house may be preferred.
-- Alok did not yet reply regarding the feasibility of running assays with other unlabeled peptides, including p53 peptide.
He needs to provide this analysis for both old and new HPLC, with emphasis on new HPLC if it is ready. If possible,
it should also be indicated whether these assays with unlabeled peptide could immediately follow the assays with labeled peptide for
honokiol/SIRT3.
AU: Yes the assay with unlabeled peptides is feasible and can be done on either HPLC. As of now I have three different peptides which will be evaluated individually. If you agree, I will start optimizations after current deadline is complete.
In addition to verification of the above, the information requested regarding basis for peptide sequences used for sirtuin kinetic assays will be required
from Alok before further feedback can be provided on the unlabeled MnSOD substrate for SIRT3/honokiol stuides.
AU: Garske and Denu (2006) say that changing peptide length from 5-8 amino acids does not affect Kcat/Km values whereas changing peptide length from 18 to 12 does not affect Kd values significantly as described by Cosgrove et al., 2006 .
Regarding use of HPLC for initial rate kinetic analysis of sirtuins, it is Alok's responsibility to review literature if needed.
I already provided example of Nat Comm paper in 2015 that used HPLC with SIRT2 initial rate kinetics. If he believes there is something
special about SIRT3, he needs to demonstrate that with appropriate arguments.
AU: As I mentioned, I have not seen a paper describing kinetic analysis of Sirt3 by HPLC. And we have to rework on the protocol to get it done. Experience from Sirt1/2 will be helpful.
===============
Fri 4/15/2016 4:58 PM
RC: Initial rates: you have looked at calculations of how much product is formed at various times.
In previous discussions you suggested that the data you have indicate that detection of product at the earlier times in initial rate experiments will not be possible.
You didn't do initial rate studies partly because of this.
You can answer based on what you know.
AU: Initial rates: So far my experiments were not design to look at the amount of product formation as a function of time. I always did endpoint assay with Sirt3. These experiments were done for 2 hours (only first experiment was done for 1 hour and since the product was low, I decided to go for 2 hours). In previous discussion, I speculated that, since even in two hours the product formation was low in that experimental condition, it will be hard to detect the initial time points. I was never asked to do initial rate studies. I started with endpoint assay.
RC: My comment about every conceivable experiment below means you shouldn't need to be asked to do initial rate expts in order to comment on their feasibility with HPLC.
You mentioned difficulty of detection in previous discussion.
Do you adhere to that view now?
And if so, how do you reconcile that with the fact that most other groups using HPLC are able to detect initial rate products?
See also below.
We need answers to both the above based on whatever info you have. One does not draw conclusions about what is possible only after doing every conceivable experiment.
RC: I also asked about your expectation about whether this will improve with new HPLC,
AU: As I mentioned earlier (before the HPLC was bought), we need a reliable and robust system. We also discussed the S/N ratio which depends upon both on HPLC, and the column. We are using lot of BSA in the reaction (which come from assay buffer supplied by Enzo), If allowed I will be interested in removing the protein from the reaction mix before loading on the column. This will improve the detection limit as there will be less material to adsorb on. E.g. In couple of days, I ran many samples, I think my column is again getting bad (or the problem with air entering in the column) as I see split in one of my peaks (as of now I am not sure what the exact problem is). These things will be minimized by new system and the column issue can be minimized by removing protein prior to loading.
RC: I would like to know if you have confidence that you will be able to run initial rate experiments with HPLC using new instrument.
I am somewhat surprised if, assuming you are not sure, you have not shared the issues you have encountered with detection of products (other peptides and initial rate expts) with tech support for these instruments, compared to literature, etc. and inquired whether you would be able to reproduce such experimental data as reported in lit with these instruments.
From: Xiangying Guan
Sent: Friday, April 15, 2016 4:24 PM
To: Raj Chakrabarti; Alok Upadhyay
Subject: RE: Draft of Proposed experiments
Please see below.
Thanks,
Xiangying
From: Raj Chakrabarti
Sent: Friday, April 15, 2016 12:23 PM
To: Xiangying Guan; Alok Upadhyay
Subject: RE: Draft of Proposed experiments
Answer to following is not sufficient:
"RC: Yes, but you checked the peak area/concentration correlations and you do have an idea of what amounts are detectable. For this assume the amount of product will be same as in FdL assays.
AU: Since I am using only FdL at this time. I did not consider looking at this aspect, even if I do, we can only assume. To get experimental data I have to do combination of experiments. Amount of product will not be same as FdL because the fluorophore contributes in to the absorption and the peak area.
Also I didn't receive answer about initial rates.
AU: I have not done initial rate experiment for any peptide. I have done only endpoint assay with only FdL.
RC: You have done various correlation studies with other peptides alone and calculations are on wiki. I asked you to assume amount/concentration of product will be the same. I didn't refer to absorbance being the same.
Based on this you can provide an answer.
AU: When I started using HPLC, I ran different p53 peptide alone (no enzyme reaction) and calculated the relationship between peak area and the concentration. Once I got that, I ran a reaction with FdL and SIrt3. In this reaction whatever I got as product and calculated the activity (as activity was less than Enzo’s reported value). Based on this, I calculated the theoretical value to see if the p53 peptide product peak will be accurately quantifiable. I need time to go back and dig out the data and refresh how it was calculated. Quantify the peaks of current experiments and then compare to see the theoretical peak area of other peptides. I will do this and upload on wiki later as I need to quantify these peaks too. Please remember, right now I am using Sirt1, not Sirt3 and they have different activities.
RC: Initial rates: you have looked at calculations of how much product is formed at various times.
In previous discussions you suggested that the data you have indicate that detection of product at the earlier times in initial rate experiments will not be possible.
You didn't do initial rate studies partly because of this.
You can answer based on what you know.
AU: Initial rates: So far my experiments were not design to look at the amount of product formation as a function of time. I always did endpoint assay with Sirt3. These experiments were done for 2 hours (only first experiment was done for 1 hour and since the product was low, I decided to go for 2 hours). In previous discussion, I speculated that, since even in two hours the product formation was low in that experimental condition, it will be hard to detect the initial time points. I was never asked to do initial rate studies. I started with endpoint assay.
We need answers to both the above based on whatever info you have. One does not draw conclusions about what is possible only after doing every conceivable experiment.
AU: It is understood that I cannot do every conceivable experiment before we draw conclusion otherwise it is waste of time. This is the reason we pick and choose different condition from published work, and broad enough to match with XG’s experiment. Even though we have certain lower substrate concentration in the plan, we start from higher side of the concentration (still sub-saturating). Since, the goal is to see if the activation occurs or not, starting from higher side (still sub-saturating) will make things clear right away. For example, if I see activation at 400 uM NAD and 100 uM peptide in 30 min, this will confirm that the activation is working for a particular modulator and we reach our goal, repeat the experiment for reproducibility, and drop others conditions and move on to next set of experiments with different modulator.
RC: I also asked about your expectation about whether this will improve with new HPLC,
AU: As I mentioned earlier (before the HPLC was bought), we need a reliable and robust system. We also discussed the S/N ratio which depends upon both on HPLC, and the column. We are using lot of BSA in the reaction (which come from assay buffer supplied by Enzo), If allowed I will be interested in removing the protein from the reaction mix before loading on the column. This will improve the detection limit as there will be less material to adsorb on. E.g. In couple of days, I ran many samples, I think my column is again getting bad (or the problem with air entering in the column) as I see split in one of my peaks (as of now I am not sure what the exact problem is). These things will be minimized by new system and the column issue can be minimized by removing protein prior to loading.
Are the controls reliable without intrinsic fluorescence?
XG: Without intrinsic fluorescence, the control is fine, as long as we taking into account of %DMSO used in reaction.
Is it possible to eliminate false positives?
XG: If the modulator without intrinsic fluorescence, the false positive will most likely be eliminated.
RC: Are you saying increase in Delta AFU in presence of DMSO is due to increase in enzyme activity not its intrinsic fluorescence?
XG: DMSO does not emit light at 460nm. However, in the previous results, the addition of DMSO do bring up the Delta AFU. Without proper standard curve, it’s hard to properly interpret if is due to increase in enzyme activity.
XG: So far, we have tried (1) adding DHP before/after incubation, (2) different order of adding developer. The only thing we do not try is have the reaction and DHP solution separate papered and incubate at the same time. Then 60mins later add developer and read out. The add the numbers together as control. Please advise if this make sense to you.
RC: You need to consider it more carefully. You don't know that the combination of DHP and deacetylated peptide produces same AFU as the sum of the two separately.
You should consider whether the use of the correct deacetylated standard (below) will allow us to do rigorous controls (comment separately on the case of intrinsic fluorescence and no intrinsic fluorescence) and suggest them accordingly.
XG: Yes, we want to know exactly what we have in the system and we must synthesize both peptides to reduce the variability
XG: We tried many times but they would not disclose the nature of standard as it is proprietary. Alok and I think, we should synthesize both (Ac-pep, and DeAc-pep with fluorophore for FdL assay) and test the reaction using their system. Theoretically it must work the only concern is, we do not know the nature of developer and its chemistry. Please suggest if we should order this, and MnSOD substrate.
RC: What does nature of developer have to do with the use of the deacetylated peptide as a standard, given that the deacetylated peptide is the product that is being formed in the reaction? In the reaction the developer is applied to deacetylated peptide product formed. Don't you want the standard to be the same as product formed in the reaction? If not, why not? Please comment.
AU/XG: Yes, we want to know exactly what we have in the system and we must synthesize both peptides to reduce the variability.
As noted you should order this.
AU/XG: We will find out who will synthesize for us as this peptide has modification and then we will request for quote.
I am waiting on Alok's answers re: MnSOD substrate.
AU: I will find out, if available, how and why they choose the length of the peptide mentioned earlier for the assay. We know that FdL peptide works in FdL assay (4 amino acids with fluorophore) and other people have done the assay with different peptide. We can get some ideas about it. I remember Ping used a peptide for modeling, what was the rationale behind using the type and length of the peptide. This will be helpful too.
Raj
From: Xiangying Guan
Sent: Friday, April 15, 2016 10:57 AM
To: Raj Chakrabarti; Alok Upadhyay
Subject: RE: Draft of Proposed experiments
Please see below.
Thanks,
Xiangying
From: Raj Chakrabarti
Sent: Thursday, April 14, 2016 5:14 PM
To: Alok Upadhyay; Xiangying Guan
Subject: Re: Draft of Proposed experiments
Last question about what batch you are using was also not answered.
Raj Chakrabarti, PhD
Chief Strategy Officer
PMC Group International
Sent from my iPhone
On Apr 14, 2016, at 5:07 PM, "Raj Chakrabarti" <[email protected]> wrote:
AU: I have not done experiment with any other peptide (so far only FdL peptides) and only end point assay. I cannot comment on if I will see or not see the product unless I do the experiment. Could you please link the above article you mentioned? Thank you
RC: Yes, but you checked the peak area/concentration correlations and you do have an idea of what amounts are detectable. For this assume the amount of product will be same as in FdL assays.
Also I didn't receive answer about initial rates, or about how things would change with new HPLC.
From: Raj Chakrabarti
Sent: Thursday, April 14, 2016 5:06 PM
To: Alok Upadhyay; Xiangying Guan
Subject: RE: Draft of Proposed experiments
You still omitted answering some of the questions below. In some places, you answered one question, and ignored the rest.
From: Alok Upadhyay
Sent: Thursday, April 14, 2016 4:58 PM
To: Raj Chakrabarti; Xiangying Guan
Subject: RE: Draft of Proposed experiments
Please see below.
Thank you
AU/XG
From: Raj Chakrabarti
Sent: Thursday, April 14, 2016 1:14 PM
To: Alok Upadhyay; Xiangying Guan
Subject: RE: Draft of Proposed experiments
See below.
Your responses are not yet satisfactory -- some of the important issues are still unclear -- and, some questions were unanswered.
Ideally you would add the commentary on why certain conditions were chosen to the experimental plan doc itself.
From: Alok Upadhyay
Sent: Thursday, April 14, 2016 11:03 AM
To: Raj Chakrabarti; Xiangying Guan
Subject: RE: Draft of Proposed experiments
Please see below.
Thank you,
Alok
From: Raj Chakrabarti
Sent: Wednesday, April 13, 2016 10:29 AM
To: Xiangying Guan; Alok Upadhyay
Subject: RE: Draft of Proposed experiments
- We discussed importance of explaining the choice of experimental conditions. Your document describes many concentrations that differ between experimental series; some of these would seem arbitrary unless explained.
- How did you choose the varying reaction times?
Short of arbitrary but within the published (Sinclair Nature paper)
RC: Not satisfactory/unclear, and you did not answer the previous question. There are many varying experimental conditions (concentrations, times), and they are not explained other than to indicate that the concentrations are nonsaturating.
AU/XG: While reading the Sinclair’s paper, Fig 1b, the maximum activation takes place around at 25 and 50 uM peptide concentrations. So we choose these two concentrations just to be sure that we see activation. For NAD, please see Fig 1c, the maximum activation takes place below 800 uM, again just to be sure we choose three different concentration and not necessarily we will try all. We start with higher NAD (400 uM) then go down to see if the activity can be detected or not. For reaction time they say 0, 5, 10, and 20 min for their study so we took 10 (to cover one time point from them) and 30 min (have extra time just in case..). The whole idea to see the activation.
RC: Still doesn't explain why your times and concentrations differ between different experimental series (enzyme, modulator).
XG: The planned conditions are based on the nature of enzyme and modulator. They are not fixed at this point. The conditions may be further modified along the outcome of the titration experiments. For example,
· We pick 10 and 30 min time points for SIRT1 but 30, and 60 min for SIRT3, that because (1) to find out if at desired time point the reaction reach the maximum (2) if the product pick is detectable by HPLC.
· Different %DMSO are used for different modulator, that because the solubility are different from one to another.
- Why are only resveratrol conditions in red?
The red highlight is to show that the concentration we picked from Sinclair paper which they highlight in their own text and the rest, we took it out from figures. Others are not in red because they did not use lower concentrations except for Honokiol.
-“We will firstly test out if DHP1c stock solution is soluble in 1% DMSO assay buffer”– you must mean resveratrol
Yes
You did experiments with resveratrol before. You need to explain the conditions and results and put any new results in that context. What DMSO concentration did you use that time? Control had no DMSO for any experiments?
Alok did not do the Resveratrol before. All the previous resveratrol results will be in one file for comparison.
RC: You didn’t answer the question regarding what DMSO concentration was used previously, and what controls were done before.
I need to understand what you are changing now.
XG: For previous Resveratrol/Sirt1 the control had 0% DMSO. Currently we are using 1%.
RC: Explain further: you didn’t need DMSO to dissolve resveratrol before, but then you found others needed to use it?
XG: In the previous experiment resveratrol was dissolved in 100% DMSO. Then diluted to final concentration in 5% DMSO. The reason why assay buffer was used as control was that for that set of experiments NAM was tested at the same time. Since NAM was prepared in assay buffer, therefore the assay buffer was applied as control. In current experiment, resveratrol was dissolved in both 1% DMSO and 5% DMSO. It’s soluble in both. Then we decided to use 1% DMSO in these experiments.
- For SIRT1/DHP1c, it appears the assumption is that the activation could only occur under the conditions where resveratrol was found to activate?
Since we tried saturating concentrations and DHPs and Honokiol did not work for some reason, and the goal of this study is to see if the activation work or not, we decided to start the study similar to Resveratrol.
- Sinclair paper did not use saturating NAD+ for any studies? Initial rate studies?
Figure 1 b and c had shown initial rate studies in Sinclair’s paper and saturating NAD+ was used in his work.
RC: You previously mentioned he did not use saturating NAD+. You meant only for endpoint assays?
So, if you found activation, would you do any follow-up studies at saturating NAD+ or are you planning to save that for later?
AU/XG: We were not clear on whether they used saturating concentration or not as we focused on getting activation done, they used saturation concentrations too when they dis initial rate studies. We will include saturating concentrations to test if we still see activation.
- By titration for HPLC, do you mean that in each case one will start without modulator and determine which conditions provide detectable product? This will differ for each experimental series, even for the same enzyme?
Do we have no information on this for either SIRT1/SIRT3 under any of the planned conditions?
RC: No answer provided.
AU: Now I know for Sirt1 for some extent (using FdL peptide). But for Sirt3, I have not done the experiment (using different concentration of NAD and different concentration of FdL peptide). The only experiment I have done is to use of different enzyme concentration with fixed substrates and fixed time. If possible and if you have such report handy, please direct me.
AU/XG: I am including modulator along with the titrations. Since we have different time points, HPLC and FdL will match at some points. For HPLC, after 10 min, the peak is not detectable (or will be difficult to quantify). Since 30 min is good enough for this study, we will stick to 30 min.
RC: Not clear. You are using the same enzyme and substrate for different series of experiments. Why can you not assess whether the product is detectable under particular conditions ([peptide],[NAD+],time) through just one series of experiments in absence of modulator?
AU: The experimental draft we have several conditions and adding modulator at the same time, we will know two things simultaneously- If product peak is detectable and if modulator is working or not. Even though we have listed several conditions, by running couple of them, we will know which one to pick from the list and move on for next experiment.
- Different conditions may eventually be used by Guan and Alok, correct?
If HPLC titration gives beyond planned, XG will cover that conditions to match the experiment.
- Importance of commentary on results, including previous results. The verbal discussion was not enough as indicated. You will need to provide detailed commentary in response to --all-- the questions in my recent emails, including the one from yesterday, and you will need to explain all discrepancies between results and methods used in previous data going back to 2014 up to now. You should expand upon points as you deem necessary - your responsibility.
XG: I am in the process of making comparison of the methods used and results obtained from previous data. In order to not to delay the progress of planned experiments, I would like to provide requested info along with the new data as the way experiments proceed. For example, currently we are working on Resveratrol/SIRT1, then the report for this part will include previous data with method used for comparison. After we wrap up Resveratrol/SIRT1, we move to DHP1c/SIRT1, by then the report will include related info at that time. And so on.
RC: I asked a number of questions to you recently by email and in meeting, including feedback on the questions you planned to ask the authors. In the latter, for example, I asked about why you think you got 2x the \Delta AFU that JMC reported, assuming that DHP 1c is an inhibitor. Did all that background come from DMSO?
XG: Do you mean, assuming that DHP1c is an activator, why I think I got 2X Delta AFU that JMC reported? In FdL assay, it’s been found that the addition DMSO will bring up the Delta AFU, which is higher than assay buffer alone. I would say some of the background come from DMSO.
When do you plan to provide these commentaries and explanation -- at the end of the month?
AU/XG: regarding author question email, since we are not going to send the question out, and we are busy at the same time, we thought it is less priority. We will try to get it done tomorrow.
RC: The questions did not have to do with the author emails. They were just listed there in some cases.
AU/XG: OK. Will do.
- What are the differences between the newly proposed experiments and previous experiments in each case, and why?
Since we tried saturating concentrations and DHPs and Honokiol did not work for some reason, and the goal of this study is to see if the activation work or not, we decided to start the study similar to Resveratrol.
RC: Ideally the doc would list what was done previously in each case and how the new conditions differ.
AU/XG: We will keep in mind. Thank you
RC: Please do so shortly.
- DHP experiments involve use of lower peptide concentration to see if peptide Km modulation plays a role. They do not appear to replicate the JMC protocols. In that case why is JMC protocol relevant?
You should also comment on how you interpret previous results of experiments under JMC conditions where the relevant controls were not always done.
We have to do the experiments to see if any condition works.
- Why is the minimum [NAD+] for honokiol lower than that for DHP?
If you look at Fig 9 of the paper, they see activation as low as 100 uM, this is the reason it is lower than DHP. And for DHP too, if those NAD doesn’t work, we might have to go further down to see what works.
RC: I mean --higher-- than that for DHP. You chose to test 25uM NAD for DHP, but only as low as 100uM for honokiol.
These numbers seem arbitrary without explanation.
RC: No answer.
AU/XG: For Honokiol the lowest concentration reported is 100uM NAD and we don’t know the peptide concentration. We can try lower/higher if 100uM not work and different combination of such. Also, we should either get full length MnSOD they used or at-least synthesize peptide, please suggest if we should get it.
- MnSOD peptide: When time permits Alok should provide some background on how he selected the previous peptides from literature and where those peptides were first used with sirtuin activity assays. We will then consider the subsequences that may be most relevant to binding and activity for MnSOD.
We agreed to synthesized following peptides-
P53 peptide- Sauve and Schramm 2003, and Hirsch et al., 2010
Pep1: NH2-KKGQSTSRHK(KAc)LMFKTEG-COOH
Pep1c: NH2-KKGQSTSRHKKLMFKTEG-COOH
H3 peptide- Landry et al 2000
Pep2: NH2-ARTKQTARKSTGG(KAc)APRKQLC-COOH
Pep2c: NH2-ARTKQTARKSTGGKAPRKQLC-COOH
K642acetylated human acetyl-coenzyme A synthetase 2 (AceCS2) used by Hirsc et al 2010 and by Ping in Plos one paper-
Pep3: NH2-TRSG(KAc)VMRRLLR-COOH
Pep3c: NH2-TRSGKVMRRLLR-COOH
RC: I also need to see commentary from the first papers that used these peptides, on how those particular subsequences were chosen given what was perceived to be minimum necessary for binding and catalysis (if available).
AU:I do not have the original articles for these. I need to look and find the original papers; it may take some time to dig it out. I will upload as it becomes available.
- p53 peptide (or any other one we have) is not being used anywhere? Please explain.
Alok has not used any other peptide in his assay yet. Only Honokiol used different protein (MnSOD), rest used FdL, we decided to use FdL peptides. The goal is to see if the activation is there or not, we decided FdL peptide will be comparable. Although we are planning to do Honokiol experiment with FdL peptide, we need to order either full length MnSOD protein or peptide flanking K122 in order to reproduce Pillai et al 2015 paper results.
RC: I would like Alok to confirm that with the current HPLC, he is incapable of detecting product accurately from any other peptide except FdL, even in endpoint assays. He also cannot detect any product for any substrate in initial rate assays, correct?
Do you expect this situation, which is in stark contrast to all published papers that use HPLC for sirtuin activity studies (see, e.g., Nat Comm 2015 on SirReal2 inhibitor kinetics) to improve with the new HPLC?
AU: I have not done experiment with any other peptide (so far only FdL peptides) and only end point assay. I cannot comment on if I will see or not see the product unless I do the experiment. Could you please link the above article you mentioned? Thank you
RC: Yes, but you checked the peak area/concentration correlations and you do have an idea of what amounts are detectable. For this assume the amount of product will be same as in FdL assays.
AU: Since I am using only FdL at this time. I did not consider looking at this aspect, even if I do, we can only assume. To get experimental data I have to do combination of experiments. Amount of product will not be same as FdL because the fluorophore contributes in to the absorption and the peak area.
Also I didn't receive answer about initial rates.
AU: I have not done initial rate experiment for any peptide. I have done only endpoint assay with only FdL.
- I'm not sure we ever discussed Alok's honokiol results to-date in any level of detail.
Was there no detectable change in activity?
This should be included in the discussion.
Alok did not post the data on wiki.
RC: Ok, why not?
AU: I did the experiment on Friday, we had meeting on Monday then we started working on these experiment. Since then the HPLC is continuously running.
But he will discuss when he presents these results. He did not see activation in the previous experiment.
- As I understand Honokiol/SIRT1 was left for later since it is not as high priority as SIRT3.
We are not trying to study Honokiol/Sirt1 in this set of experiment. We are trying to see if can reproduce someone else’s work and if so in what conditions. So Honokiol/Sirt1 will be done or decided to be done after these studies are over, based on these results.
- In the FdL experiments, there is no mention of the controls. These need to all be planned out carefully.
See our previous discussions.
Provide the reasoning behind the controls.
Or are you assuming that some FdL controls are less relevant due to use of HPLC as well?
The AFU signals from the experiments (NAD + Peptide + Assay buffer + DHP1c in 5%DMSO) were measured. The results show that the AFU readout (2063) of DHP1c solution after 60min incubation at 37oC decreased comparing to 0min (2382), which indicated the AFU signal provided by DHP1c solution was not stable over the incubation time. The results also indicated that the AFU signal from control experiments (add DHP1c fresh after 60 min reaction) was not comparable to AFU data from those experiments (add DHP1c at the beginning of the reaction and incubate 60min at 37oC).
The use of HPLC eliminates such problem due to intrinsic fluorescence.
RC: You still didn't state what controls you plan to run and why.
XG: OK. I got your point. In FdL experiments, the controls will be used are listed below
Resveratrol/SIRT1: 1% DMSO
DHP1c/SIRT1: 5% DMSO
Honokiol/SIRT3: 5% DMSO
DHP1c/SIRT3: 5% DMSO
As discussed this is very important -- since in the past you used, e.g., controls without DMSO...Have you concluded that adding DHP1c fresh after 60 min reaction and then waiting another 60 minutes before measurement would not be a useful control?
XG: After addition of developer(containing 2mM NAM solution), the reaction in principle will be quenched and fluorescence signal release from deacetylated peptid will be readout. 15-45 min will be the range in which the readout is stable, after that the AFU will go down. Therefore, the control like “adding DHP1c fresh after 60 min reaction and then waiting another 60 minutes before measurement” will not present the accurate readout.
In summary, what is your conclusion about the possibility of developing proper FdL controls?
XG: So far, we have tried (1) adding DHP before/after incubation, (2) different order of adding developer. The only thing we do not try is have the reaction and DHP solution separate papered and incubate at the same time. Then 60mins later add developer and read out. The add the numbers together as control. Please advise if this make sense to you.
Are the controls reliable without intrinsic fluorescence?
XG: Without intrinsic fluorescence, the control is fine, as long as we taking into account of %DMSO used in reaction.
Is it possibleto eliminate false positives?
XG: If the modulator without intrinsic fluorescence, the false positive will most likely be eliminated.
If not, are other non-HPLC based assays preferable for screening? Which?
XG: I need to study on this. Will get back to you early next week.
We cannot keep using an assay if it is not reproducible. We cannot avoid this issue.
See also questions about the deacetylated standard.
XG: We need to synthesize the deacetylated/acetylated peptide as standard.
Also, I didn't see the table you presented at meeting posted anywhere.
XG: It’s been posted under Task list from lab/DHP1c intrinsic fluorescence control
- On a related note, have you ever considered ordering deacetylated FdL peptide for use as a standard?
Alok has mentioned this before but for some reason, we did not get OK from you.
RC: As I recall, it was not just a matter of not giving an OK. We had discussed it in context of running a standard curve for HPLC, but Alok
said he did not use that method for quantification (rather, he used Denu's method). Alok subsequently provided evidence suggesting that he had identified
the product peak, and did not indicate ordering the deacetylated FdL peptide was a priority.
Here I am also referring to its use in FdL assays. Why do we rely on "deacetylated standard" for our standard curves, given
that they didn't tell you what it is? Please comment regarding whether they claim it is proprietary. Why wouldn't we just use the
actual deacetylated labeled peptide for the FdL standard curves? If we do, what are the remaining sources of variability? Comment.
We should get it now.
AU/XG: We will find out the sequence and send it out for synthesis. We should also get either MnSOD protein or the peptide for honokiol experiment.
RC: Didn't get answers to questions above.
XG: We tried many times but they would not disclose the nature of standard as it is proprietary. Alok and I think, we should synthesize both (Ac-pep, and DeAc-pep with fluorophore for FdL assay) and test the reaction using their system. Theoretically it must work the only concern is, we do not know the nature of developer and its chemistry. Please suggest if we should order this, and MnSOD substrate.
- Do you have bulk SIRT3 now?
Not yet
RC: Then what batch are you now using?
XG: We received two SIRT1 kits late last week and yesterday. SIRT1 lot number is 02161501 with 5U/ul. We almost used up one kit.
When are SIRT3/1 bulk orders coming in?
AU/XG: We will email Enzo and update accordingly.
- Note that you will need to take the responsibility of planning your work such that you can finish whatever experiments you consider relevant to reaching the conclusion of whether these compounds activate, within the specified time we discussed.
Post to wiki as needed.
We will upload the data and these discussions on wiki together.
From: Xiangying Guan
Sent: Tuesday, April 12, 2016 11:19 AM
To: Raj Chakrabarti; Alok Upadhyay
Subject: Draft of Proposed experiments
Dr Raj
Please check the attached draft of proposed experiments. Please provide feedback if the proposed experimental plan is scientifically designed. Please comment on if the proposed experiments is sufficient to serve our goal.
Thanks,
Alok and Xiangying
AU: 4-4-2016: Experiment AU15- Intrinsic fluorescence mesurements of Honokiol.
I used difference concentrations of Honokiol in assay buffer with 5% DMSO to determine the fluorescence of the compound.
Excitation wavelength 355 nm
Emmision was measured from 330-600 nm.
I will upload the data tomorrow but it appears that there is no fluorescence activity around 460 nm which is used as readout in FdL assay.
AU: 4-4-2016: Schedule for the week 4-4-2016-4-8-2016
1: Experiment AU15-Determine the intrinsic fluorescence of the Honokiol- Monday (4-4-2016) Update (4-6-2016): AU15-Honokiol fluorescence.xls
2: Experiment AU16-Determine the activation of Enzo Sirt3 by DHP1c following XG's reaction conditions- Tuesday (4-5-2016) Update (4-6-2016): AU16b.xls
3: Experiment AU17- Determine the activation of Enzo Sirt1 by DHP1c following XG's reaction conditions- Wed (4-6-2016): Update (4-6-2016): Experiment will be done tomorrow (4-7-2016) AU17-4-7-2016.xls
RC (4/4/16): Honokiol EC1.5 by HPLC, following up on XG's FdL studies with honokiol, will need to be scheduled as well.
4: Experiment AU18: Determine the activation of Enzo Sirt3 by Honokiol- Thursday ( 4-7-2016), conditions will be based on XG's experiment. Update (4-6-2016): Experiment is will be done on Friday (4-8-2016)
AU: 4-8-2016: I am still running the samples on HPLC and I will analyze and upload the data on Monday.
AU: 4-4-2016: Experiment AU14- Honokiol Solubility.
AU14-Honokiol solubility.docx
Email Question from Raj-
"Please consult with XG as needed and get feedback. I previously discussed w her and we posted data on wiki. I'm hoping you two communicated and you read that posting before starting your work."
"Note that DMSO has an issue in this regard.
You should also discuss this in that context."
"Have we confirmed lack of significant intrinsic fluorescence?"
AU: I will discuss with XG regarding DMS and solubility issue.
AU: 4-4-2016: Experiment AU13- Sirt3/DHP1c experiment-
AU13-3-30-2016.ppt
AU: 3-28-2016
Experiment AU12- NAM concentration vs peak area determination.
AU12-3-25-2016.ppt AU12-3-25-2016.xls
AU: 3-25-2016
Experiment AU11- Reaction with 75 uM DHP2c
AU11-3-24-2016.pptAU11-3-24-2016.xls
AU: 3-24-2016
Experiment AU10- NAM dose response:
[NAD+] = 500 uM, [FdL peptide] = 100 uM, Enzo Sirt3 = 1uL, Time = 60 min, Temp. = 37 degree C.AU10-3-22-2016.pptAU10-3-22-2016.xls
AU: 3-28-2016 NAM IC50 update.
By looking at the data, it appears that the IC50 will be in between 10-15 uM. I need more data points to be sure.

AU-21-2016
Experiment AU8: AU8-3-16-2016.ppt
The effect of DMSO (1 and 5%) on HPLC chromatogram compared to no DMSO. This was done to see the if DMSO affects/overlaps with reactant peak just by looking at the retention time. It seems that DMSO will mask O-acetyl-ADP-Ribose and affect NAD+ peak. There is. in general, slight baseline change when DMSO is used.
Experiment AU9: AU9-3-16-2016.ppt AU9-3-16-2016.xls
The DHP2c dose response curve in presence of 5% DMSO. The DHP2c concentration used were 25, 50, 100, 150, 300, and 500 uM, [NAD+] = 1.5 mM, [FdL Peptide] = 100 uM.
The reaction was carried out at 37 degree for 2 hours.
Some of the observations can be made, such as- For some reason, the DHP2c unable to activate in this set of reactions, % product formation is pretty consistent in the reaction, and 5% DMSO does not inhibit enzyme.
I also discussed about DHP1c with XG but time did not allow me to start the reaction. Should we move to DHP1c, or try some variation in DHP2c experiment to see if it is working or not? Please let me know so that I can plan the experiments on Monday. I will be available via email, if you have any question.
=
AU: 3-16-2016
Experiment AU4- Testing DHP2c dose response on Enzo Sirt3 (New vial of enzyme)-
AU4-Dose response curve.ppt
AU4 data-3-11-2016.xls
Experiment AU6: ADP-Ribose retention time
AU6-ADP-Ribose.ppt
AU6-ADP-Ribose.xls
Experiment AU7: Enzyme concentration v/s product peak area calculation-
AU7-3-15-2016.ppt
AU7 data-3-15-2016.xls
AU: 3-9-2016
Following observations can be made -
Respective peaks are there regardless whether TFA or DHP2c is present or not. In this set of the reactions, I don’t see disappearance of the peak. Is this reproducible? I have to repeat the experiment to confirm this. So, why I did not get the peak last time? I am not sure, may be the column is much cleaner this time. One thing can be easily seen that the high noise level in the peak area disappeared too in this experiment. Although the column is clean, the pressure fluctuation is still present, which can be seen in some other peak’s shape (not properly shaped).AU3-troubleshooting.pptAU3-troubleshooting-3-9-2016.xls
AU: 3-3-2016:
Priority experiment list (see the email communication below):
The column will be ready tomorrow afternoon (I need extensive washing with the buffers I am using to run actual samples, as of now I am washing the column with 2-propanol-water, the buffer I use for experimental runs are acetonitrile and water), and I will try to run a control sample to see if everything is fine. If everything is fine with column, the following experiments which you prioritized will be done by Monday, at-least one time.
1 c) Reaction without DHP2c, but add DHP2c (10uM) after stopping the reaction
RC: Priority, though not a solution. This is just meant to confirm that DHP 2c is not in any way interfering with the reaction, and that the problem is caused by DHP in the detection step, by verifying we also have no detectable peak if the reaction is carried out without DHP.
2e) With DHP2c incubated for 2 hours (No TFA)
RC: Priorities, if you can do these in a reproducible way without quenching the reaction. I am not aware of the lead time for running HPLC experiments and the variability of that,
so I cannot comment on the reproducibility.
3b): 400-500 pmole of deacetylated standard + 10 uM DHP2c incubate for 2 hrs at 37 degree
RC: Priority
4e): 400-500 pmole of deacetylated standard + 10 uM DHP2c incubate for 2 hrs at 37 degree (no TFA)
RC: Priority.
====
AU: Enzo’s deacetylated standard may not behave in similar fashion as we do not have clear idea what it is.
RC: Agreed. However, if the properties are similar, the standard curve expts above would reveal if there is any prospect of getting a signal despite dhp precipitation/TFA, without adjustment of pH (expt 3b) and the extent to which dhp precipitation/TFA reduces the peak area of standard/product (expt 4e). I expected you would run a curve (i.e., more than one concentration of standard), but if you are confident that 400-500 pmol should be enough, that is fine.
AU: Running a standard curve will take full day (4-5 concentration) which can be done once the priority experiments are completed at-least once. The 400-500 pmoles I choose because this will be close to the product formed, I don’t want to go lower because I want to see a peak even after ~20% less absorption, and I don’t want to go too high either.
====
Email communication with Raj on Wednesday, March 2nd, 2016
Dear Dr. Chakrabarti,
I will do two experiments in parallel to identify the problem and see if we are headed in the right direction. This is very comprehensive list of experiments but I think, things will be much clear once I finish Set 1and 2. I may not have to do set 3 and 4.
Set 1: Reactions with 2uL of enzyme;
a) Without DHP2c incubated for 2 hours (control experiment)
b) With DHP2c incubated for 2 hours (to see if time is affecting the peak, I already know this but it is prudent to include this)
c) Reaction without DHP2c, but add DHP2c (10uM) after stopping the reaction
Set 2: Reactions with 2uL of enzyme- this will show if TFA (acidic environment is playing a role); I assume that after two hours of incubation, the enzyme will become too week to carry out any significant deacetylation.
d) Without DHP2c incubated for 2 hours (No TFA)
e) With DHP2c incubated for 2 hours (No TFA)
f) Reaction without DHP2c, but add DHP2c (10uM) after stopping the reaction (No TFA)
Set 3: Incubating DHP2c with Enzo’s deacetylated standard, to see if DHP2c has similar effect-
a): 400-500 pmole of deacetylated standard incubate for 2 hrs at 37 degree
b): 400-500 pmole of deacetylated standard + 10 uM DHP2c incubate for 2 hrs at 37 degree
c) 400-500 pmole of deacetylated standard incubate for 2 hrs at 37 degree, add DHP2c after adding TFA
Set 4: Incubating DHP2c with Enzo’s deacetylated standard, to see if DHP2c has similar effect, and if TFA is involved too
d): 400-500 pmole of deacetylated standard incubate for 2 hrs at 37 degree (no TFA)
e): 400-500 pmole of deacetylated standard + 10 uM DHP2c incubate for 2 hrs at 37 degree (no TFA)
f) 400-500 pmole of deacetylated standard incubate for 2 hrs at 37 degree, add DHP2c (No TFA)
Enzo’s deacetylated standard may not behave in similar fashion as we do not have clear idea what it is.
I am still cleaning the column; it is really a slow process as the pressure is going very high so I have to reduce the flow rate to 0.3ml/min. It is taking longer time than expected.
I am looking into the protocols related to AE purification where authors used strategy to remove chaperon from the target protein. Once it is ready, I will update you.
Thank you,
Alok
Reply by Raj via email on March 2nd, 2016:
RC: -- Yes, as you noted yesterday, reduction in solubility of DHP 2c due to low pH may be causing the problem. This is consistent with what we know about DHP 2c's solubility properties.
-- Please see below. I have highlighted the priority experiments since there is a timeliness issue.
-- Please add the plan with rough dates to wiki. Ideally within a few days we will have an Hplc dose response curve since this problem may not be too difficult to resolve.
When will column be ready?
[Email continue below, more in-line responses from RC]
I will do two experiments in parallel to identify the problem and see if we are headed in the right direction. This is very comprehensive list of experiments but I think, things will be much clear once I finish Set 1and 2. I may not have to do set 3 and 4.
Set 1: Reactions with 2uL of enzyme;
a) Without DHP2c incubated for 2 hours (control experiment)
b) With DHP2c incubated for 2 hours (to see if time is affecting the peak, I already know this but it is prudent to include this)
RC: I thought the above two were already done, so they are of lower priority if there is any issue with conducting and analyzing all experiments in parallel.
c) Reaction without DHP2c, but add DHP2c (10uM) after stopping the reaction
RC: Priority, though not a solution. This is just meant to confirm that DHP 2c is not in any way interfering with the reaction, and that the problem is caused by DHP in the detection step, by verifying we also have no detectable peak if the reaction is carried out without DHP.
Set 2: Reactions with 2uL of enzyme- this will show if TFA (acidic environment is playing a role); I assume that after two hours of incubation, the enzyme will become too week to carry out any significant deacetylation.
d) Without DHP2c incubated for 2 hours (No TFA)
e) With DHP2c incubated for 2 hours (No TFA)
RC: Priorities, if you can do these in a reproducible way without quenching the reaction. I am not aware of the lead time for running HPLC experiments and the variability of that,
so I cannot comment on the reproducibility.
RC: If we confirm that DHP solubility / TFA was causing the problem, or if there are issues with reproducibility of the above, there are couple of other approaches to 2e) that might be important to render the results more reproducible:
-- neutralization of pH of solution (eg w/ NaOH) after TFA deactivation of enzyme (assuming TFA irreversibly deactivates enzyme).
(In fact neutralizing pH after irreversible enzyme deactivation might separately help with other issues like resolving ADPR peak since we know it is discernible in absence of TFA. This can be assessed later after more careful consideration.)
-- alternatively any method of deactivating enzyme without altering pH (hence retaining DHP 2c solubility) could be used (we discussed approaches like NAM).
f) Reaction without DHP2c, but add DHP2c (10uM) (No TFA).
RC: Lower priority.
Set 3: Incubating DHP2c with Enzo’s deacetylated standard, to see if DHP2c has similar effect-
a): 400-500 pmole of deacetylated standard incubate for 2 hrs at 37 degree
RC: Somewhat lower priority (after 3b and 4e), since goal is to see if we can detect any peak, not the change in peak area with respect to the absence of DHP.
b): 400-500 pmole of deacetylated standard + 10 uM DHP2c incubate for 2 hrs at 37 degree
RC: Priority
c) 400-500 pmole of deacetylated standard incubate for 2 hrs at 37 degree, add DHP2c after adding TFA
RC: Probably not required at all.
Set 4: Incubating DHP2c with Enzo’s deacetylated standard, to see if DHP2c has similar effect, and if TFA is involved too
d): 400-500 pmole of deacetylated standard incubate for 2 hrs at 37 degree (no TFA)
RC: Lower priority.
e): 400-500 pmole of deacetylated standard + 10 uM DHP2c incubate for 2 hrs at 37 degree (no TFA)
RC: Priority.
f) 400-500 pmole of deacetylated standard incubate for 2 hrs at 37 degree, add DHP2c (No TFA)
RC: Probably not required at all.
Enzo’s deacetylated standard may not behave in similar fashion as we do not have clear idea what it is.
RC: Agreed. However, if the properties are similar, the standard curve expts above would reveal if there is any prospect of getting a signal despite dhp precipitation/TFA, without adjustment of pH (expt 3b) and the extent to which dhp precipitation/TFA reduces the peak area of standard/product (expt 4e).I expected you would run a curve (i.e., more than one concentration of standard), but if you are confident that 400-500 pmol should be enough, that is fine.
AU: 3-1-2016: Task #7 update-
Dose response curve for DHP2c-
Enzo Sirt3 = 2 µL/rxn
FdL peptide = 100 µM
[NAD+] = 1.5 mM
[DHP2c] = 0, 10, 25, 50, and 75 µM
Reaction volume = 50 µL
Reaction time = 2 hours at 37 degree Celsius
Blanks-
Blank 2- All components, no Enzyme
Blank 3- All components + 50 µM DHP2c, no enzyme (concentration is arbitrary, it could have been any from 10-75 uM).
Two major points came out of this experiment-
1: The retention time difference between Ac-Peptide and product (DeAc-pep) is about 2 minutes whereas the retention time difference between Ac-pep and deacetylated standard is about 1 min. As mentioned earlier we do not know the nature of deacetylated standard provided by Enzo. They would not disclose it.
2: The enzyme reaction without DHP2c works (at least, I see a peak as compared to the blanks suggesting the presence of DeAc-peptide). The problem is when I add DHP2c; I don’t see the “DeAc-pep” peaks. This happened in all the reactions where I used DHP2c. As of now, I am not sure what is causing the “disappearance” of product peak from DHP2c experiments. We need to discuss it further. I am unable to come up with a testable hypothesis except that DHP2c may be insoluble and try doing the experiments directly in minimum % of DMSO allowed. Even this explanation does not explain why a product peak will disappear. The following observations can be clearly made using chromatograms-
1: The baseline is very noisy in the gradient area whenever I used DHP2c. This may suggest some sort of degradation interfering in product peak resolution (Its just pure speculation).
2: The overall absorption (Area) is decreased about 20% whenever I used DHP2c even in the blank as compared to Blank w/o DHP2c or reactions w/o DHP2c.
AU2-2-26-2016.pptAU2-2-26-2016.xls
Since I have Guanidine-HCL, I am going to clean the column.
======
AU: 2-16-2016 Updated schedule-
Revised Task Schedule-Alok 2-12-2016.doc
AU: 2-16-2016:
Two quick point I want to reemphasize-
1: I planning to use 12U Sirt3 for each reaction, and 100 uM DHP-2c will be done after I complete reaction with 10, 25, 50, and 75 uM.
2: Regarding increasing the reaction volume, even if I go to 100 ul reaction volume, keeping all the component concentration same, there is really small space to include DHP2c for endpoint studies, unless I get better activity Urea purified enzyme.
RC: Alok,I don't have time to revisit these points with you at this time due to other responsibilities. I posted extensively earlier and we discussed.
You stated that we can go above 100uL (no limit) if needed.
AU: Yes, I agreed to 100 uL reaction volume, I just wanted to be upfront in saying that DHP2c has its solubility issue in buffer.
AU: XG and I discussed to see what is the minimum total volume needed for the reaction using 10U (0.1U/ul) in-house enzyme with only 25 uM DHP2c. This comes out as 150 uL.

There are many pending tasks that were not yet completed and the schedule also does not properly reflect all recent discussions and emails. I will not repeat them all here. In particular,
you did not account for the experiments with in-house enzyme and altered reaction volume. The schedule mentions p53 but does not indicate anything about volume or
the enzyme that will be used, and the fact that FdL might be used instead of p53, the basis upon which the choice of substrate will be made, etc.
All these points have been mentioned in our correspondence and wiki.
AU: I thought I will do a quick 100 uL reaction to see if it works then update the schedule accordingly.
AU: Since in task 7, i am using Enzo Sirt3 and FdL peptide to do dose response curve, task 8c is essentially same except peptide. If 150 ul reaction volume works, then I can use in-house enzyme for these reactions (substrate p53). If not then I will use Enzo Sirt3.
The reaction in 150 uL is for 25 uM DHP2c. If I want to go higher say 50 uM DHP2c then I have to change the reaction volume again. Since I will use minimum 100 uL enzyme (assuming 10U total; 0.1U/uL), and one purification batch generally gives 2 ml, I can do 15-16 reactions in one batch.
Not clear what you mean by concentrated enzyme availability, and there is not time to discuss now.
AU: "concentrated enzyme availability" comment was made before our discussion (I wrote on 2-12).
I am going to forward all my emails to you to XG for her to review. She will discuss
with you and give me feedback on her impression of what you are saying now given our previous discussions.
AU: 2-12-2016: Updated task schedule- [2-17-2016 date for task #7 is tentative, will depend upon concentrated enzyme availability, I will update the schedule accordingly again]
AU: 2-12-2016
Protein purification schedule (using Urea method)-
Since we had bacterial pellets in the -80, Sudipto was quick to purify one batch on Friday (1st batch). We have enough cells to do one more batch which he will use on Monday, and protein will be ready by Wednesday EOD. If everything is comparable to previous purifications, XG can use this batch for her experiments. I will use 1st batch but the concentration is low. I need 8-10 U/reaction. Sudipto can try to concentrate 3 ml to less than 1 ml to get about 4ug/ul final (as of now the concentration of the protein is not known). If loss of activity is seen again, we will not concentrate the protein in future. To continue my experiments, I will use Enzo enzyme for task 7, 8.
Sudipto, please see if you have any feedback and comment accordingly.
Thank you
AU: 2-9-2016:
Email from Raj-
Following up on convo from yesterday:
Calculations 1:
-- compare Enzo activity as obtained from HPLC to that from fdl. If not consistent, indicate this immediately.
AU: 2-9-2016:
If we look at the Enzo Sirt3:
I used 8U of Enzo Sirt3 enzyme
By Enzo, one unit is defined as amount of enzyme producing 1pmoles/min.
By this, theoretically my reaction should produce = 4pmoles/min x 120 min = 480 pmoles total
In my reaction, I used total 8 units for 2hrs. Theoretically, I should have gotten 960 pmoles of the product, but I got 188 pmoles/min (EDIT-AU: 188 pmoles total, not per minutes). We have to keep in mind this reduction in efficiency while running the reaction.
So, the actual product formation is 5.1 times less than Enzo’s claim.
RC: Alok, I have reorganized your answers since they mixed together purified enzyme and Enzo.
More importantly, when I asked about Enzo, I was not asking about Enzo's quote on activity, but rather the activity measured by FdL in our lab.
It seems this was actually partly answered below instead. You should make a direct comparison between your result in pmol and Guan's.
AU: 2-10-2016: I will get this once she is free.
It seems Guan's result is consistent with Enzo's quote?
But why are we quoting results from Nov 2015? Was that the same Enzo batch? Didn't we just do another Enzo control experiment with FdL when we were working with EC1.5
of DHP 2c?
AU: I will ask her to look at this concern and reply accordingly.
XG: EC1.5 of DHP2c was tested for purified enzyme. The data provided was the most recent EC1.5 (DHP1c) measurement with Enzo, when we got new batch of DHP1c from Santa Cruz.
As I understand, your assessment of pmol product formed in your HPLC studies was based on Enzo's quoted concentration of the "deacetylated standard"
through your concentration-peak area correlation (i.e., an HPLC standard curve).
AU: I did not run the standard curve on HPLC, I followed Borra and Denu (2003) method for quantification.
RC: Ok, this assumes the absorbance properties of acetylated and deacetylated FdL peptides are identical. This may be quite accurate.
However, we also need to be aware that the background is quite significant and we may not be able to rule out differences in background contributing to
quantification error. We will get a better idea about that as we run more background studies.
Both FdL and HPLC standard curves use the same deacetylated standard.
Also, reactions for FdL and HPLC use the same substrate. FdL relies on the assumption that deacetylated standard and deacetylated peptide product have the same AFU/uM
relationship, whereas HPLC relies on AU/uM relationship being the same.
Please confirm.
AU: For FdL assay, yes it is correct that deacetylated standard and deacetylated peptide product will have the same AFU/uM but for HPLC, it should be Area/uM if a standard curve is used for quantitation.
RC: That's fine, I was referring to units on the amplitude. We have also done some studies with peak area in the case of FdL.
The important point is the relevant assumptions regarding the correlations between spectroscopic properties and concentration for the deacetylated standard and acetylated product being the same.
Since we don't know what the deacetylated standard is, it may not have the same absorbance properties as the deacetylated product.
By this token it is good to use the Bora + Denu method rather than standard curve for quantification in HPLC, as you did.
The fluorescence properties may match more closely, since that is what the standard was developed for (still, there may be differences between AFU from the standard and product).
I will consider the results after receiving answers above. Relative comparison of Enzo activity with / without DHP2c will be needed in meantime.
Regarding your last email:
"I used 8U of Enzo Sirt3 enzyme. Enzo defines 1U enzyme is the enzyme producing 1pmole/min at 37 degree.
In my reaction, I used total 8 units for 2hrs. Theoretically, I should have gotten 960 pmoles of the product, but I got 188 pmoles/min. We have to keep in mind this reduction in efficiency while running the reaction."
Please ask Guan to comment on whether she saw a similar phenomenon with Enzo and Fdl when she quantified product with standard curve.
AU: 2-9-2016: XG, please see the comment, last paragraph, from Raj
XG (2.9.16): The calculation is listed below. Please be aware the use of proper standard curve is important for calculation.
 |
| pmole calculation.GIF |
-- report specific activity and purity of Arctic Express
AU: 2-9-2016 Arctic Expressed Sirt3 (in-house) purity and specific activity-
Purity: 40-45% purity specific activity (0.92191U/ug)- purified once!
The extra protein band eluting with the target protein ~70kDa, because of this the overall purity is low. If this extra band is not there, the purity will be >80%
RC: So the purity is currently significantly lower than either Enzo or urea.
It was mentioned purity was higher. Is this the best result obtained so far with Arctic Express?
What was the latest plan for removal of the extra band (next steps we discussed re: chaperonin) and how long would it take.
As you know time is limited for the current study.
AU: 2-10-2016:
This data in powerpoint was emailed to you on October 9th, 2015 explaining purity and specific activity of different enzymes we had that time. Protein purification_10.9.2015.pptx
RC: Yes, I'm referring to our discussion this week when I believe it was indicated that Arctic Express had higher purity and activity.
Please reply regarding the plan questions above.
AU: Sorry, I was preoccupied with HPLC issues and posted partially.
When we were discussing purity of the different preparations, I said: purity of the enzymes are in following order "Urea > Arctic Express> Enzo", what I meant that Arctic Express without Chaperon band. My apologies, I wasn't explicit about this.
When I was doing Arctic Express, I tried incubating the enzyme with ATP/Mg/KCl mix to remove the Cpn60. I did one experiment but did not work at that time. I do not think this method is applicable to all the protein, although they authors claim that. Next I was thinking to incubate the lysate with sub optimal level of Urea, say 1-2 M, to see if this will free the Sirt3. Soon after one group published the same method claiming 2M Urea releases Chaperon. Also, they mention that ATP/Mg/KCl strategy did not work in their hands.
Sudipto is suggesting another method, incubating Arctic Express lysate containing Sirt3 with heat inactivated bacterial lysate to remove the Chaperon.
I would say that we should drop the ATP/Mg/KCl idea and focus on Suboptimal Urea and heat inactivated bacterial lysate and try them simultaneously. I would say 2-3 weeks to see if these strategies will work.
-- calculate predicted deacetylated peptide and ADPR coproduct peak areas (2 hrs) for Arctic Express (fdl,p53 - latter assuming same activity. p53 to be done after expt below)
-- calculate predicted deacetylated peptide and ADPR coproduct peak areas (2 hrs) for urea (fdl,p53 - latter assuming same activity. p53 to be done after expt below)
Urea,fdl peptide: Sudipto told me that he did FdL assay twice with his batch 2 enzyme. This is the batch I used for my reactions. He got 0.06U/ug and 0.1U/ug relative specific activity.
Theoretical product formation calculation-
Let’s assume 0.1U/ug is correct and I used 20 uL (20ug) and 40 uL (40ug) total protein in the reaction.
The highest amount of Enzyme I took is = 40ug x 0.1U/ug = 4U
If I follow same calculation, in-house enzyme will produce 94 pmoles (480/5.1). If this is true, then it is too low and similar to the Blank reaction.
RC: Waiting on these calculations.
AU: 2-10-2016:
Assuming p53 peptide will behave similar to the FdL peptide, and assuming equivalent activity, say 1 U is taken for the reaction and 2 hrs reaction time; following predictions can be made for the product in two hours of reaction regardless of source of enzyme used, be it Enzo, arctic expressed or Urea purified.
In 120 min., 120 pmoles of each product will form.
Since I do not have lower concentration, I am calculating with 4000 pmoles DeAc-enzo-peptide I ran twice; one 4000 pmoles alone and another 4000 pmoles in combination with 4000 pmoles Ac-Enzo-peptide.
So 120 pmoles of DeAc-Enzo peptide will give about 310350 (or 318225) area.
For 120 pmoles of ADPR, area will be 91391 (91391, 95914, 93855, calculated from 4000 and two runs of 10000 pmoles)
For 120 pmoles p53 peptide, the area will be 98951 (calculated from 400 pmoles, single run). I need to run higher pmoles to see if numbers are comparable).
I will revisit these numbers tomorrow and update if they change, after stringent quantification of the peaks.
Please let me know if I understood the question correctly and feedback if further information needed.
RC: You'll need to edit these when the p53 peak area/concentration correlations are in. When do you expect to have that? What is the status of HPLC?
Please also answer the questions below about minimum detectable area for each peak.
AU: 2-12-2016: The 200 pmoles of DeAc-p53 peptide quantification is not very accurate. When I compare 400 pmol/200 pmol area, the ratio is 2.4. Theoretically, it should be 2. I need to run 600, 800, and 1000 pmoles to get accurate area values.
AU: 2-11-2016
Scott is tentatively scheduled to fix the HPLC on Tuesday. I will do p53 peak-area correlation day after he fixes it.
AU: 2-12-2016- If he finishes early, I will try to run 600, 800, and 1000 pmoles DeAc-p53.
Of course, we may need to use more than 1U, especially for initial rate experiments. The amount needed will depend on the peak used for quantification and its background/minimum detectable area.
The important issue that has been pending is this: you found (see above) that undetectability of urea enzyme product was consistent with fact that only 4U was used. Why did you use 4U instead of 8U, as you did with Enzo? I assume because you did not check with Sudipto. It seems you chose the amount based on a prespecified volume rather than comparable activity. I did not get a clear picture of what the limitations imposed by volume are (if any), and have been planning to inquire, since by simply doubling the U, you may be able to detect the product. Given urea enzyme specific activity, it appears you need around 80ug of urea enzyme per reaction to achieve the U that you are finding is the minimum required for accurate detection using deacetylated peptide peak (please verify this under Calculations 2 below). How much urea enzyme do we have left? Is it enough for all planned experiments? Are there any issues with the volume of enzyme solution required per reaction?
Related, what is specific activity of Enzo and how does it compare to urea enzyme? (Since specific activity is probably not available, compare U/uL instead).
If there are no limiting factors regarding reaction volume and amount of available enzyme for the purpose of the proposed experiments (the ones I recently listed below), it's not clear why there would be any interest in considering the use of Arctic Express for the purpose of our current experiments, especially given the significantly lower purity of the latter. Low specific activity may in itself be undesirable, but this is not specific to HPLC and will be discussed separately.
AU: 2-11-2016
In 50 uL reaction, the maximum volume of Urea purified enzyme I could use was 40 uL which equals to 4U. Please remember, Sudipto did assay twice, same enzyme, he got two different values 0.06 and 0.1 U/ug. I don't think we have enough Urea purified enzyme, Sudipto needs to purify few more batches as soon as possible. The current Enzo Sirt3 specific activity is 10.4U/ug. I consulted Sudipto to see the specific activity before the start of my experiment.
Also, XG should comment on how the concentration of product for initial time points in initial rate experiments compared to that at endpoint, in order to roughly determine the U of enzyme needed for initial rate experiments with HPLC, given the minimum detectable peak areas. I understand this will likely be quite high, but it is useful to have a rough idea. If too much is needed for initial rate experiments, then we may still use urea enzyme for endpoint experiments.
If we have enough urea enzyme for all planned experiments and there are no issues with reaction volume, please update the schedule and proceed with the experiment proposed below to verify you can detect appropriate U of urea enzyme product in endpoint study, as the priority.
AU: 2-11-2016
Given the current specific activity of urea purified enzyme, volume is the limiting factor. Also, we need to purify more enzyme.
AU: 2-12-2016 Technical discussion-
Assuming the specific activity of urea purified enzymes remains same (0.06 or 0.1 U/ug). I can use maximum 40 uL. My reaction components are as follows-
5 uL NAD+ (final 1.5 mM)
5 uL Ac-Peptide (100 uM)
40 uL Enzyme (~25 uM) : Just a reminder, this 40 uL is equal to 4U of enzyme which is unable to produce detectable signal.
Even if this combination works, there is no space for DHP2c. Since maximum amount of DHP2c which can be dissolved in buffer is 100 uM, for 10, 25 uM DHP2c final, I need 5 or 12.5 uL (of 100 uM DHP2c) respectively. The only way this can be circumvented to to high activity enzyme. Or please share other ideas.
Expt (one day):
-- check p53 in isolation; would not be comparable to fdl, hence not useful to do too many initial
rate expts, but could be used for Km,kcat w/,w/o 25uM DHP 2c
--Verify that the signal would be more significant and detectable than fdl peptide with urea
(through calculations 1 above)
RC: These should be done by midday I believe.
AU: 2-10-2016:
We have some problem with HPLC, it is leaking, injector misalignment, and I am trying to fix it with the Scott's ( The person who came to fix HPLC earlier) help. Issues started this morning.
Meanwhile I ran p53 peptide alone yesterday to see the detection limit for deacetylated peptide.
Following concentration were run-
Ac-Peptide-
400, 800 pmoles
DeAc-peptide-
400, 200, and 100 pmoles
I have not quantified them yet, but 100 pmoles look similar to background (Buffer alone). Today I wanted to run the 4000 pmoles of Ac-Peptide to see if there is any peak appearing corresponding to the DeAc-peptide position.
Calculations 2:
-- I understand repeating the experiments (or better, running lower concentrations of each isolated component) will eventually be required to properly assess signal to noise and integration accuracy,
but nonetheless: provide your best current estimate of the minimum concentration of deacetylated peptide and ADPR coproduct that
can be detected (for ADPR assume no overlap with void peak -- assume NAM quenching with no change in background at that wavelength;
for deacetylated peptide assume the background peak).
You have run isolated components at low concentration so you should have an idea of the minimum peak area and hence concentration of each that can be accurately quantified.
RC: This should be possible to complete within the next day or so while HPLC's are running.
Also, provide the approximate # of enzyme U required to achieve these minimum peak areas (2 hrs).
AU: 2-12-2016
For minimum peak area determination, enzyme is not needed.
Further experiments:
-- Assuming there was some consistency between Enzo activity obtained from HPLC and that from fdl: run Enzo with 25uM DHP, compare to fdl. If earlier expts were not consistent, move to next steps below where we attempt to improve quantification of product peaks.
RC: If enough urea enzyme is available for all the proposed experiments with HPLC and there are no issues with using more enzyme per reaction, we should use urea enzyme instead of Enzo for all the experiments.
The priority should be to demonstrate that urea enzyme product is detectable with the same U previously detected for Enzo, as predicted.
-- proceed with dose-response for urea enzyme, DHP in assay buffer as planned [revise the schedule: by how many days will completion of this be delayed compared to current schedule? RC will check
after proposed schedule is posted]
Assuming urea enzyme product is detectable via HPLC using fdl or p53 substrate and we have enough enzyme, no issues with respect to volume of enzyme solution used:
-- Use urea enzyme with fdl or p53 (since results with urea enzyme may be comparable to Guan's, may prefer to use fdl) to do DHP dose response, possibly in 1% DMSO.
-- Assuming initial rate experiments products can be detected, then do kcat,Km for latter system via HPLC (fdl is again preferable; some experiments will be repeated with p53).
Subsequently, as time permits, proceed with other approaches discussed to enable better HPLC quantification of initial rate products, settle the minimum concentration of each product that can be quantified, and comment on possible improvements with another instrument/column.
Please post your replies on the wiki, under each of the above tasks, as the results are in. Also, please post all the Q&A from yesterday on the wiki as well. Answers from Guan and Sudipto should already be available, and some of the above calculations which we discussed should be quick.
Raj
===========
AU: 2-9-2016
Email from Raj dated Feb 8th, 2016:
Alok,
You need to clarify some points below in detail before I can reply.
-- you are referring to background of the deacetylated peak in the absence of reaction, which you speculated may increase with acetylated peptide concentration, correct?
AU: Yes
-- you need to explain 200uM. From what I can surmise you mean that if you did 200uM, you would get an idea of whether the deacetylated background is increasing linearly with [peptide]?
In the ppt you said you ran just acetylated peptide already, but I assume you mean you did not run it with other reaction components other than enzyme.
AU: The peptide substrate alone I ran was 100 uM concentration in assay buffer. None of the other components were present, not even with enzyme. It was just to see the relative position of the peaks.
-- regarding titrating 25-100uM, I am not sure what you are referring to; if you wanted to increase enzyme activity due to low sensitivity, you might consider going higher, but it seems you are suggesting going lower.
AU: As of now, it appears that 100 uM is saturating (as 200 uM is giving similar values. Since I did not run the background for 200 uM, I cannot say it for sure but if I assume the background is linear with peptide substrate concentration, theoretically it should be twice as of 100 uM peptide substrate. If so then the % deacetylation readings will be 4.7% and 3.8% for 100 uM and 200 uM respectively (for this calculation, I just doubled the background concentration and substracted from 200 uM reaction product).
-- However, the [peptide] was supposed to be chosen to be saturating (was this 100uM in your experiment?). This is relevant to the focus of our study.
to increase activity, we may consider endpoint assays at different [NAD+] (this would not apply to initial rate studies), but that would need to be considered carefully.
Please verify with XG that peptide was saturating in this particular reaction and if not, let me know why it was not since it should be in the FdL assays.
AU: I will confirm with XG what peptide concentration is saturating in FdL assay.
Separately, I have several points to communicate regarding your latest results vis-a-vis next steps and I will be sending them shortly in an email.
I would like you to review these points, provide preliminary feedback by email and then we can discuss as needed. This will expedite discussion.
Raj
==
Regarding titrating 25-100uM, I didn't see the answer on the questino regarding the reason for that proposed experiment. You replied regarding 200uM and 100uM but not 25-100.
Do you mean to increase signal to background ratio by decreasing [peptide]?
Regarding titrating 25-100uM, I didn't see the answer on the questino regarding the reason for that proposed experiment. You replied regarding 200uM and 100uM but not 25-100.
AU: Oh, sorry.
Since I do not know, as of now, what is the minimum saturating concentration in HPLC assay that is why I was talking about it. If 100 uM come as saturating concentration, I will use that.
This was easiest way to confirm.
Raj
=====
AU: 2-9-2016 Email from Raj and discussion:
Alok,
Please answer the following, which will simplify discussion:
1) Void peak / ADPR coproduct peak separation
a) Does void peak location depend on our specific instrument/column?
AU: Void peak retention time mostly dependent upon column properties, and length/volume. A specific instrument (HPLC) may place a role e.g. reproducibility of retention time. Other than this, void peak is determined by column.
b) If so could this have lead to different overlap with ADPR coproduct for Sauve?
c) Have you explored modification of retention times to separate the peaks (void peak/ADPR peak)? Is it more likely that Sauve achieved separation this way?
AU: Since Sauve did not mention any detail, like column length, id, solvent condition, separation gradient etc, my answer is very speculative that yes, he may have achieved separating ADPR manipulating these parameters. I needed more time to resolve this issue, since I have dead-line to meet, I did try only two things and seems did not work. I changed the flow rate, and initial isocratic run time to see if I can resolve the issue.
2) ADPR coproduct peak
a) Can we check whether the amount of oaadpr formed for the specified level of activity of Enzo enzyme would be detectable in absence of void peak , based on our adpr Hplc studies?
I believe you effectively ran a "standard curve" for ADPR already, so you should be able to answer this.
AU: The void peak will be there even I just run buffer (peak area will be smaller, time remains same). I am not sure how to avoid this initial peak. I did run ADPR standard earlier (without TFA), but as I mentioned once I add TFA, both peaks are merging.
3) Validation of FdL with HPLC for Enzo enzyme
a) You should now be able compare your quantified Enzo perfect peaks w FdL results on same endpoint assays and report for validation, without further experiments?
Or do you need to run further background studies? If so, propose the latter.
If this is possible now, it is a priority.
AU: If time permits, I would like to repeat the same experiment to see the variation in HPLC assay. As far as comparison is concerned, am I comparing the activity of the same enzyme with two different assay system? Moreover, I think FdL assay is done for one hours whereas, HPLC assay reaction time is two hrs. Nonetheless, pmoles/min product can be compared.
4) Purified enzyme activity and Hplc sensitivity
a) Please indicate sensitivity of the Hplc assay for deacetylated product
AU: I have to run different concentrations of deacetylated product to evaluate the sensitivity (minimum amount of deacetylated peptide detected in this assay).
b) Please indicate, given what we know about the activity or purified enzyme from FdL and the HPLC background, whether we expect the product peak in your ppt from Sat for this enzyme to be undetectable (as observed).
AU: I am not exactly sure about the question, but do you mean if the product is there (by in-house enzyme) but can't be detected due to sensitivity? If so, I do not see any peak higher than the blank peak (please see the calculation slide from Sat.).
5) p53 peptide
a) Could p53 peptide provide a better signal w purified enzyme?
b) Do you have any preliminary data here? How long would it take to get it if not?
AU: 5a-b
I do not have data with p53 so I am not sure if signal to background will be different in this case. We have to do the experiment.
6) Arctic Express purification
a) Regarding arctic express purification: please summarize the purity and specific activity and compare to the others (enzo, urea)
AU: The purity of the arctic expressed protein and urea is much greater than the Enzo enzyme. I will dig out the gel pictures, and ask XG to fill in activity data as she was the one who did activity assay. I was purifying the proteins with different condition and she was doing activity assay.
7) HPLC sensitivity
a) Is there any reason to believe that the new HPLC we were considering purchasing would have better sensitivity?
This would need to be quantified and one would need to assess based on what we know about product peak areas for given concentrations studied, whether the sensitivity would be sufficient.
AU: The strongest case for new HPLC would be that, new model will be more stable in terms of detector, pump functionality etc.
Raj
Do you mean to increase signal to background ratio by decreasing [peptide]?
AU: This may be interesting possibility, I never thought of it. Thank you.
Btw, in FdL is reaction stopped by NAM?
We should review requirement for TFA quenching if it is responsible for peak merging.
The answer to ADPR sensitivity/quantification question is relevant in this regard.
AU: Yes, in FdL assay, the reaction is stopped by NAM. But for HPLC, the TFA is used because it is compatible with mobile phase used for separation. Regarding ADPR, unless we solve the initial peak problem, I cannot say it for 100% surety. If the product formation is stoichiometric, then I see ~188 pmol total DeAc-peptde, I can assume that ADPR will be in similar amount. If so, I have to independently run these reaction (without TFA) to see if this can ve seen as peak but again, we cannot avoid TFA.
Thank you
AU: After discussion on Feb8th afternoon:
Dear Dr. Chakrabarti,
Three experiments are in priority as we just discussed-
1: Endpoint assay with p53 peptide (both Enzo and in-house Sirt3)
2: Get the FdL endpoint assay data from XG and compare with HPLC data (pmol/min comparison)
3: DHP2c activation reaction with Enzo Sirt3
I will start p53 experiment from tomorrow.
I will get a detailed schedule for this and send it to you.
Thank you,
Alok
=
AU: 2-8-2016:
Revised Task Schedule-Alok 2-2-2016.doc HPLC quantification data 2-6-2016.pptx
AU: 2-2-2016
Revised task schedule : Revised Task Schedule-Alok.doc
AU: 2-2-2016
Updated task scheduleTask Schedule-Alok.doc, please comment.
AU: 2-2-2016
Email from Raj on Feb 1, 2016 and response-
1) As discussed, the dose-response endpoint studies are the priority for HPLC comparison to FdL, not initial rates.
I described the required endpoint experiments in previous emails.
As I mentioned in recent emails as well, you should be posting relevant correspondence
regarding tasks to the wiki periodically. Please do so for the recent correspondence.
2) The following HPLC experiments need to be finished before the end of February.
a) HPLC dose-response curve for DHP2c in assay buffer
b) HPLC dose-response curve for DHP2c in 0.2% DMSO (including saturating DHP); check with XG on the % DMSO.
c) Selected HPLC endpoint data for p53 peptide (for example, for a dose-response curve in assay buffer; similar time required as a)). Details will be finalized after a,b) but time should be allocated now.
AU: I have human p53 peptide corresponding to amino acids 372-389 (H2N-KKGQSTSRHK-AcK-LMFKTEG-COOH).
d) Selected HPLC initial rate data (e.g., MM parameters Km, vmax with,without 25uM DHP2c)
Some of these data will go in SI of PNAS paper. d) may go in the main text.
You should propose asap a schedule on the wiki detailing how a-c) will be done, task by task, with dates listed.
AU: I will discuss with XG regarding the concentration of DHP2c and the percent of DMSO she used in her experiments. Should I assume that I will be using Enzo enzyme for all these experiments, and Enzo peptide for a, c, d? Please feedback.
RC: No, purified enzyme and FdL peptide for a,b,d. p53 for c. I will let you know if there are any edits to this plan.
AU: Then I have to run the trial experiment with in-house purified enzyme to see if it is similar to the Enzo enzyme or I just start experiments and modify as and when needed?
You should leave at least one week thereafter for d). Hence a-c) should be done before the end of the third week of Feb.
We will specify the details of d) shortly.
AU: I ran the experiment only once, and speculating on the deacetylated peptide. Since, we do not have non-acetylated Enzo peptide, I proposed that I should run another two reactions with different concentrations of the substrate to see if the amount of deacetylated product increases. This will confirm two things; one, it will show the consistency (reproducibility) of the reaction or any variation; 2, confirm that the peak is actually a deacetylated product. If I want to quantify ADP-Ribose, I must separate this from initial peak. Keeping this in mind, I will try my best to finish these experiments within your deadline.
Sherry will discuss with you how to arrange your working hours, if needed, so as to be able to meet these deadlines.
AU: Sherry asked me to work 2 extra hours (total). These two hours can be used either lunch time or after 5. I told her that once I start the experiment, I will use in two consecutive days. This way I can extend my sample run time.
3) Regarding HPLC peak quantification, I'm not sure what you meant by manual vs automatic, but I am going to ask
Sudipto to look into quantification (not for your data, but for generic data), starting today or tomorrow first thing, so it does not take another 1-2 days.
AU: When we run a program and the software identifies a peak, the peak data such as peak start and end points, peak area etc calculated by default program, this will be automatic integration. Sometimes, the peak start and end is way off from the actual start and end. So we have to manually shift these parameters to fit. Sudipto’s help is much appreciated in this regard. The only thing is that if I am running a program, then he cannot work on the computer.
You should aim to finish the points I mentioned in my email below (required before you can start on a-d) above) within 2 working days from the time you start (since Sudipto will be helping with quantification). Please post that short term
schedule for this week on the wiki as well.
RC email Jan 29th-
--Ideally by Mon we should have settled whether we can detect coproduct or deacetylated peptide and shortly thereafter whether we can quantify them.
--By Tues we should aim to have the peak area / concentration correlations and quantified product peaks for the purpose of comparing to the analogous FdL experiment.
4) Regarding which peaks to use in monitoring product formation, I thought prior literature may have used ADPR coproduct peak for this purpose. If so, please review and comment on why we may not be able to use it.
It appears you are planning to focus on NAM. Depending on sensitivity of our instrument, I agree that we may need to tweak certain concentrations in the reaction to get measurable levels of products/coproducts.
AU: Yes, theoretically both ADP-Ribose and deacetylated peptides can be used for this purpose. I do not have a reference where they quantified ADPR for kinetics. I will search for the reference and discuss with XG if she has it handy (or if you have the reference, please direct me to the source, it will save some time for me). Again, my guess is that small ADP-Ribose peak is merging with initial peak and that is why I can’t see. As I said earlier, I may have to slightly modify the program to separate them.My focus is to get any product, the only criteria is that the peak is clear, well separated from the rest. As of now, I can see only deacetylated peptide. I proposed an experiment to confirm this notion that the peak I am labelling as deacetylated peptide is actually a deacetylated peptide. As of no, I cannot say for sure because we do not have deacetylated enzo peptide.
RC: Ok, we can focus on deacetylated peptide with increased concentration as noted above to start.
Regarding ADPR for kinetics, it is not clear whether they used it for quantifying rates, but I believe Sauve's paper indicated that they quantified that peak.
Just bear in mind one thing: that you may need to find the deacetylated peptide peaks substrate-by-substrate if we change the peptide in the future, e.g. for p53.
This would not be the case for coproduct(s), which would be one of the advantages thereof.
5) As noted you should start making ppt reports periodically and posting on the wiki (at least once a wk) under the relatedsubtasks.
AU: I will post data if I finish one set of experiments on wiki.
For today, you should proceed with your experiments as priority along with answers to the above and wiki posting; you can add existing HPLC raw data to Fri ppt if needed to clarify prior work, but focus on new experiments.
You can then send the more detailed ppt report including the product/coproduct peak identification analysis and quantified product peaks according to above schedule.
AU: I will post raw data from Friday by the end of the day today.
Raj
=
AU: 1-27-2016:I will be running FdL peptide and Enzo Sirt3 separately on HPLC to see the resolution in the current HPLC program. Lat, if everything goes well, I will run a sample reaction using Enzo substrate and enzyme using following condition as XG. XG, please comment-
100 uM peptide substrate
1.5 mM NAD+
5U Sirt3
Assay buffer to make total volume 50 uL.
Reaction will be started with the addition of enzyme, Reaction will be carried out for 1 hours at 37 degree C. After one hours, raction will be stopped by addition of 1% TFA (final).
Please comment and feedback, if this plan lacks anything.
Thanks
Alok
AU: 1/8/2016:
Updated schedule- everything remains same except #3 which I updated to 1-2 weeks. I need to do this again since we changed the injector. Task Schedule-Alok-radioactive.doc
AU: 1/7/2016:
Since HPLC was down, I was working with TopCount. I did run two C14 NAM samples to get the CPM count but I will repeat it. Mike was here yesterday to fix some software issue in the instrument. I am planning to redo the samples.
Hopefully, HPLC will be fixed today as Scott is on his way for maintenance. I will be with him and also working on TopCount. I will update schedule tomorrow.
-
RC (12/26):
- Please provide an update on status of Hplc assay and schedule MM experiments for deacetylation w the p53 substrate based on measurement of AADPR coproduct peak in early Jan if possible.
- According to schedule you are apparently currently in the assay development stage of task list
(4-6 wks).
- We need to know whether you will be able to finish this in early Jan and be able to obtain MM kinetics in presence and absence of ~ 25uM dhp 2c by mid Jan for purposes of validating the FdL assay results for this paper.
- We need three sets of MM expts - one for just DMSO as well
Saturating will not be needed
- Enzyme (diff batch) to come from Sudipto's current work with you on purification if needed. See posting on Guan's page, where we indicate that we may need more enzyme for her experiments as well. Please consult and advise.
AU: 12/29/2015
We have some problem with the HPLC (there is a leak) which will be fixed on 7th Jan. Since I already ran individual components of the reaction including products, I will run the mix of samples again to confirm the separation and retention time.
Right no, I am working with the TopCount which I think working fine. I purchaged special plates which I will use for counting. I am waiting for one more item, which will be delivered tomorrow, then I will do a simple read with C14 sample.
Because of the instrument problem, I am slightly running behind schedule but I am confident that by the end of Jan, I will have some preliminary data for deacetylation assay using Sudipto's enzyme.
AU: 12/10/2015
RC via email (12/07/2015):
We need to be aware of some differences between our approach and kinetic models and those of Sauve/Jackson, especially if those experiments take 2 months as described.
Note we will not only be using this data to determine k5 in the terminology mentioned below.
You should review w Alok the concentration combinations you do in your current experiments if you didn't already done so and then he should provide a time estimate for that approach as well. Then after I see the time required for that we will discuss modifications to the schedule / protocol as needed.
AU: As I understand, so we will also study deacetylation kinetics, inhibition of deacetylation by NAM, and activation by DHP1c using HPLC method.To this end, I am developing a method which will be suitable to study mentioned parameters by HPLC. XG and I discussed about her experimental procedure in which she uses Tecan for readout. She mentioned that she used following concentrations for her studies-
[NAD+] = 375, 750, 1500, 3000 uM
[NAM] = 0, 50, 100 uM
Time-points: 0, 5, 10, 20, 30, 60, 120 min.
[DHP1c] : 0, 25 uM
She also mentioned that it takes about 2-3 weeks to complete the experiments using above parameters. In my own schedule (point #6), I included, except DHP1c, these under assay development and predicted to be completed in 4-6 weeks. The reason I need more time is because HPLC run will take longer as compared to Tecan which is a high-throughput system. The base exchange kinetics comes after this optimization. The number of time points is similar (5 in mine 7 in XGs) my experimental design and XG's but I am not sure if I can calculate kinetic parameter (k5) using only three NAM concentrations. I can try and if I get good data then we move on.
RC: Thanks. We will need to talk in detail before settling the protocol for the "production run" experiments. As you know, we are writing papers on how to determine kinetic parameters using such experiments. (The previous authors did not develop the kinetic models that we have developed, and we are using HPLC experiments to study additional properties. Some aspects of our protocol, e.g. in terms of the concentrations needed, will be determined in conjunction with the model. I will advise on this later.)
For now, I need data on the time required for the experiments. The reason I asked for a comparison of HPLC schedule to Guan's under the same assumptions is the longer time required for HPLC analysis. It appears time time required for a single concentration combination in HPLC is about 2x that for FdL.
AU: As of now, single run needs 65 mins. This is really preliminary since I am still in optimization phase. This will likely be reduced to 40-45 min in future once I have all the components in the single mix.
We will likely change the number of concentrations of each substrate after getting all this information.
Also, we may prioritize certain experiments after receiving the feedback from reviewers on the current paper.
Alok should let me know once has verified linearity of detector in desired concentration range for the peaks of interest and then move to improving peak separation.
AU: So far I have been running individual components of the deacetylation reactions such as NAD, NAM, ADP-Ribose, and peptide to see the concentration range, effective separation (retention time). I have finished NAD, NAM, and ADP-Ribose. I am in the process of running peptide to see where it stands. Once I finish all these then I will run a mix of all these components and see if they separate well. For linearilty, I have to run few more concentrations to correlate. In my HPLC program,
NAM: 100, 200, 600 uM gives clear peak around 13.6-14.5 min (600 uM is saturating).
NAD+: 10, 30, 50 uM gives clear peak around 9.95-10.45 min (50 uM is saturating).
ADP-Ribose: 10, 50, 100 uM gives clear peak around 5-5.4 min (10 uM is too small to distinguish between noise).
These samples were run only once and are still in the optimization process. I will start peptide today and tomorrow and see if they separate well in current HPLC program.
RC via email (12/06/2015)
Please do look into the issue of hydrophobic residue at position+1 for our current and new substrates.
For example I am wondering whether for our current paper we are using a substrate that has this property or not since this property is conducive to allosteric activation.
This may be mentioned on our response to the Plos paper reviewers - you can ask guan about that.
AU: 12/11/2015:
In the current assay, XG is using same Fluor de
lys
assay system which contains amino acids 317-320 of human p53 (Gln-Pro-Lys-LysAc). This peptide is tagged with
fluorophore
. In this system, the tagged fluorophore may act like hydrophobic residue, required for the activation.
The peptide I synthesized, have either Leu, Ala, or Val at +1 position. Although these amino acids are, to some extent, have hydrophobic properties but certainly not as strong as Phe, Tyr or Try. These peptides may not exhibit hydrophobicity-dependent activation by small molecules. It will be interesting and imperative to use a previously published compound such as resveratrol (or any other compound) as a control.
The peptide sequences I synthesized-
Pep1: NH2-KKGQSTSRHK(KAc)LMFKTEG-COOH
Pep2: NH2-ARTKQTARKSTGG(KAc)APRKQLC-COOH
Pep3: NH2-TRSG(KAc)VMRRLLR-COOH
RC: Please ignore the comments regarding the FdL peptide /reviewer comments in PLOS since the fluorophore is terminal and it appears from your posting above that this is not one of the synthesized peptides.
However, there was a peptide mentioned in our PLOS article that we used for computational studies, and I believe Guan had synthesized this peptide as well (without a fluorophore). Was this the p53 peptide
upon which FdL peptide is based (as a subsequence)? This is only relevant if you are using the same peptide in any of the new experiments. E.g., is one of your new peptides the same as the one used for computational studies?
Is peptide 1 p53 peptide? It appears it may be a different subsequence that does not include amino acids 317-320.
AU: Pep3, I synthesized is same which was used by Ping for modelling in Plos paper.
Separately, and more importantly, you need to look at the paper I sent you on proteomics of SIRT substrates and check what types of hydrophobic residues are conducive to allosteric activation.
In addition to the +1 position, that paper may have noted other positions that are relevant for allosteric activation (+6?). You might check those as well.
AU: I will look into it if they have discussed hydrophobicity and allosteric activation relationship and update accordingly.
AU: The acetylome paper (Rauh et al., 2012) describes only what type of amino acids are preffered at particular position and how specificity/selectivity varies across Sirt1-7. e.g.
"Sirt1 prefers polar, mainly positively charged residues surrounding the deacetylation site (Fig. 2b), with non-charged residues inserted at positions +2, +3 and -2."
OR
"Sirt3 prefers positively charged and disfavours negatively charged residues downstream of the deacetylation site and—less pronounced—in several upstream positions.."
This paper doesn't mention/discuss the relationship between hydrophobicity and activation, I will search the original papers and see if I can find it.
RC: I have reviewed some of the data in the literature. It appears p53 peptide is not allosterically activated and that Leu +1 is not likely to display the hydrophobicity necessary for allosteric activation.
The peptides above are likely good candidates for studying non-allosteric activation mechanisms, but do check the other residue positions that may be relevant vis-a-vis the aforementioned paper.
We will do detailed kinetic studies on one of the substrates, and then show lack of substrate selectivity of the activation mechanism through endpoint experiments only on the others.
=====
AU: 11/30/2015 Updated task schedule- Task Schedule-Alok-radioactive.doc
AU: 11-25-2015:
HPLC Updates-
The problem with autosampler needle was fixed by TRITECH engineer and working ok now.
I have been trying to use the new column to see if this is working fine. As of now, it seems that the column and the HPLC unit is working fine. I am trying to run few components of the reaction mix e.g. NAD, NAM separately to see how they resolve on the C18 column. The base exchange protocol, I will be following in my future experiments was published earlier but they do not have clear protocol to resolve the reactant and product components.Right now I am trying to get a good resolution of the peaks and optimizing the buffer conditions. From next week, I will start mixture of the components and optimize the protocol for the HPLC.
I am still waiting for the fraction collector, we ordered from the GIM (UPDATE- received fraction collector just now).
RC: Ok - please provide an updated schedule (extending that below) now that the HPLC is working. Please provide a schedule for the next 2-3 weeks including the details of what you will be doing by date.
Please note that we may have need for this assay shortly, potentially within the context of the current paper upon receiving referee feedback (the experiments would need to start before that, even before paper submission).
AU: If it is OK, I will update the schedule on Monday, I will be connecting fraction collector, and some housekeeping work with HPLC such as cleaning column etc. Please suggest.
RC: Ok, please remember to do so.
AU: 11-13-2015: HPLC Update, Sherry contacted a local company (Triad Scientific) for a technical support. Maybe on Monday someone will come and fix the problem.
AU: 11-13-2015
I am trying to set up the Beckman HPLC in the radioactive room. Transferred, and cleaned all the components, and connected them to the computer, and power supply. XG and I jointly tested the HPLC pump which seems working fine. We tried to get rid of air bubble present in the tubing. During test-run/system check, I encountered a problem with autosampler. It seems that a component of the autosampler i.e. needle is not positioning itself properly thus unable to proceed further.
I contacted technical support (DeWayne Towsend) to find out the possible reason and rectification. He replied saying that the sensor controlling needle might not be working. Here is the email reply-
[I think you have a sensor problem that is preventing the autosampler from selecting the correct location of the tray and/or needle. Usually this sort of problem can be fixed by cleaing the sensor with a squirt of water followed by a blast of canned air. The most likely problem is the sensor just to the right of the needle when the needle is home. Removal of the small vial holders will expose it. The other sensor, syringe position, requires removal of the panel on the left side to gain access.
I am not sure I understand what you mean by “the system would not stop.” Does the syringe keep flushing solvent through the needle or does the sample tray keep turning? A little more detail on what happens when you click on the “Wash” would be helpful.
Beckman has stopped providing parts for the HPLC systems so if a part is broken the odds of being able to replace it are not very good. We would be glad to look at the autosampler if you want to ship it to us. We do have some parts, but we cannot guarantee we will be able to repair it.
Sorry I do not have better news.
DeWayne Townsend
Analytical Instruments.]
I updated him with more information and screen shots and waiting for his reply.
Here is some of the screen shots- Autosampler-1.bmp Wash-injector2.bmp
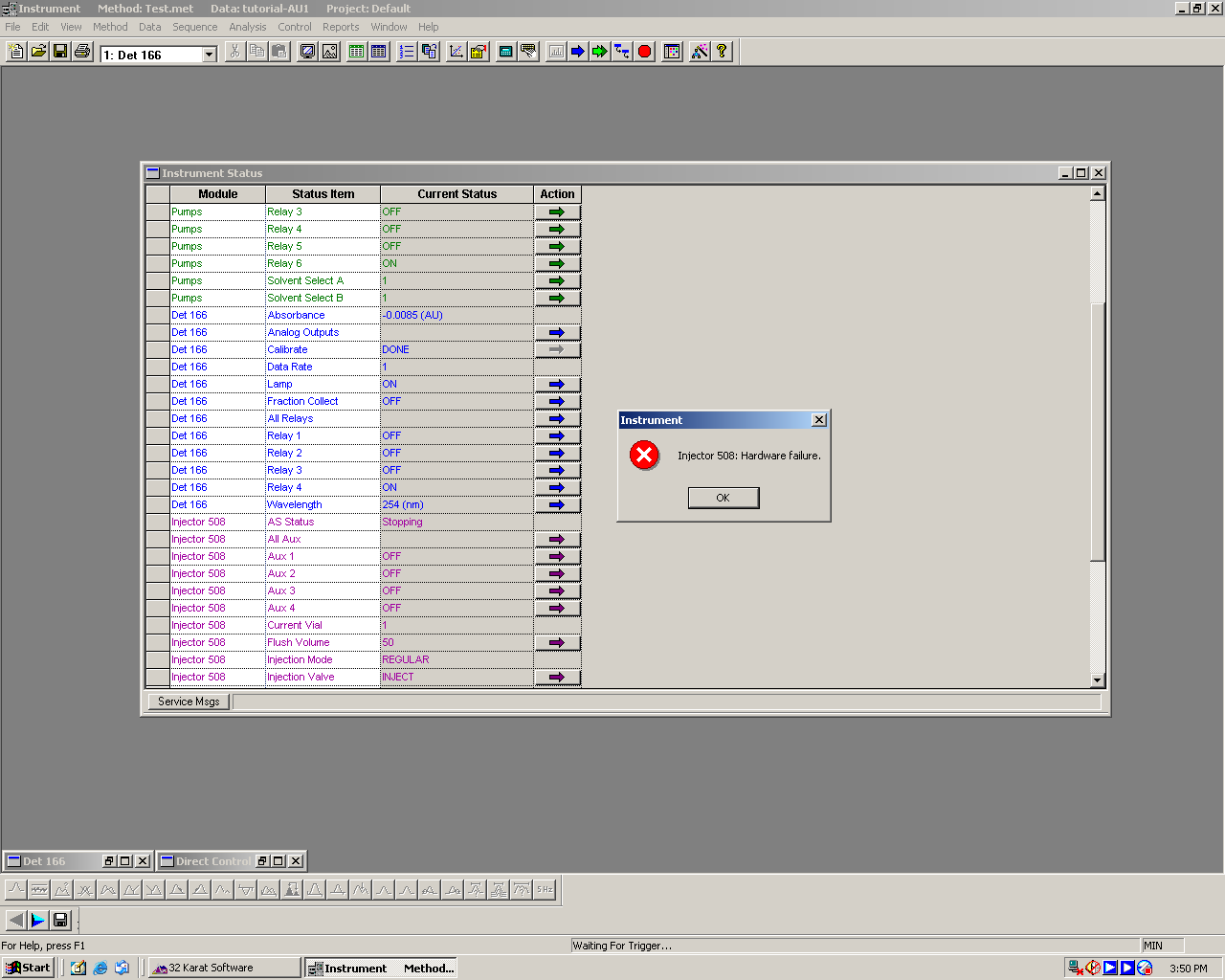{kind=link}
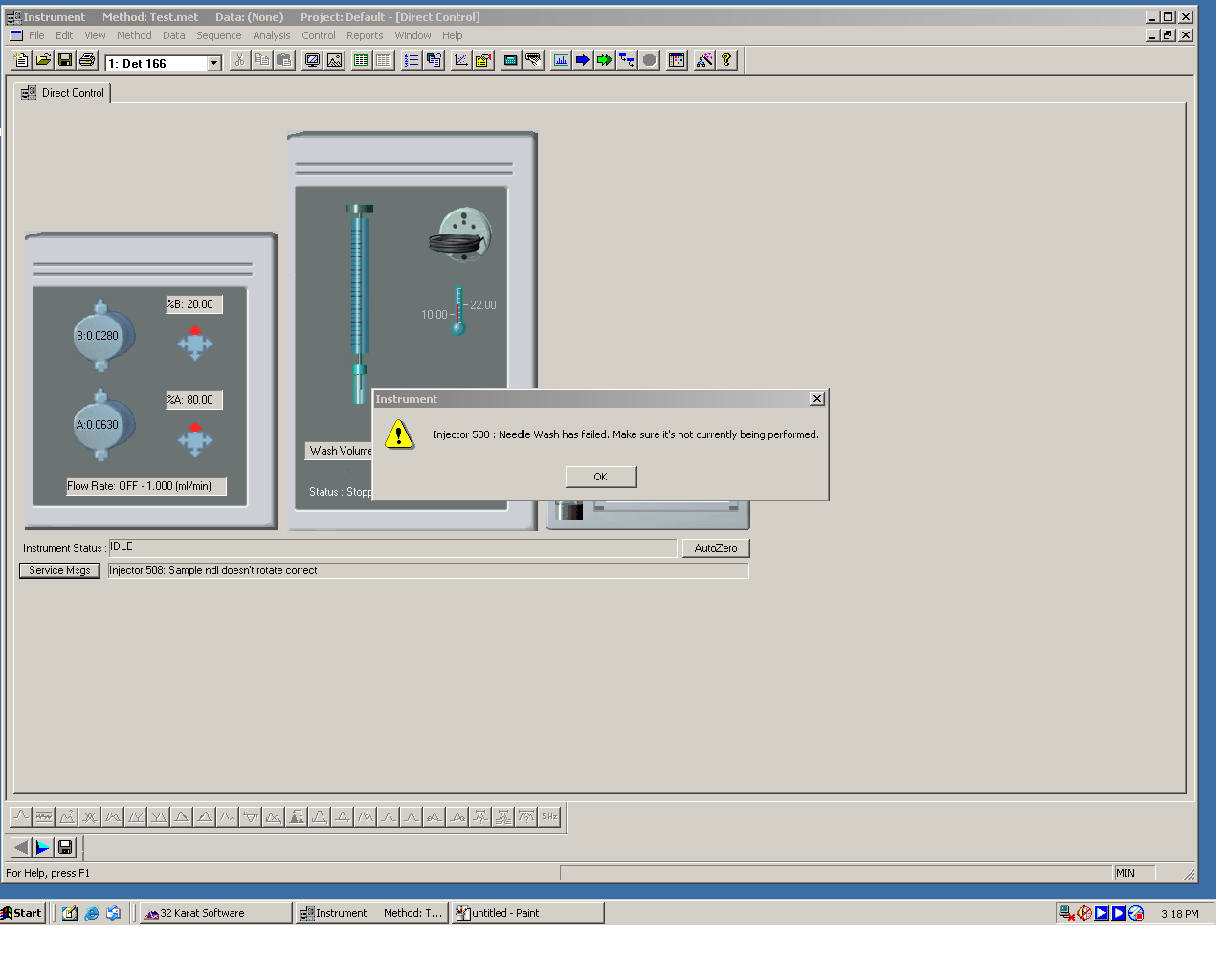{kind=link}
AU 11-6-2015: UpdatedTask Schedule-Alok-radioactive_RC_comments.doc
RC: Have you provided the detailed schedule for the last box in the table?
AU: 11-10-2015:
I haven't finalized the schedule for 7b. This step is dependent upon task #6 where I propose to optimize the reaction condition etc. This step will tell me exactly how much time I will take to run a single reaction with [NAM] = 0 concentration or any variable of this such as including more radiolabeled NAM.
Here are some variables I have to consider which will know from task #6-
1: Optimization of reaction conditions such as -
a) Reaction time
b) concentration of substrates e.g. peptide, NAD
c) concentration of NAM and its behavior/effect on reaction product.
Although above is published but we do need time to get it working on our hand which will be done in task #6.
RC: Please note that we have already chosen many of these variables per our other experiments (Guan's). See the protocol there. Please assume that we will eventually do the same types of series of experiments
(same [NAD+],[NAM], after the initial validation of the protocol for a single concentration).
There are choices to be made regarding how to combine various concentrations in this series in a single day. You should discuss this with Guan and see if changes are required for the radioactive experiments.
Peptide should be at saturating concentration.
2: Then we move to HPLC separation of the product, although this will overlap with reaction condition optimization, but we need to consider following-
a) Optimize the clear separation of the products to calculate the peak area etc
b) Optimize the amount of radioactive product to be used to get high signal/noise data.
I would like to emphasize again that the task 7b is dependent upon task #6 but a hypothetical and ideal situation will be like this for one experiment-
1: planning and starting a reaction may take 1-2 hrs
2: reaction time 2 hours (published) + 30 min to prepare the sample for HPLC (during this time I will start HPLC and get it ready for the run)
3: The limiting step is run on HPLC, as of now I am not sure if I can store the reaction mix at -20 degreeC while I am running the HPLC. It might take around 2-4 hours to run HPLC from start to finish i.e. ready for next run.
4: analyze the samples on top count, maybe 1-2 hours (?).
RC: Regarding choice of substrate peptide one thing we need to know is whether the ones we are planning to use have the hydrophobic residue that is necessary for SIRT1 allosteric activation or not.
RC: Difference between this and last file not clear. Further info is needed on question AU6 if you need feedback.
AU: 11/4/2015
Radioactive experiment tentative schedule.
RC comments added:
Task Schedule-Alok-radioactive_RC_comments.doc
RC: Please replace EC1.5 in doc above with saturating.
AU: 11-5-2015 update file with comments Task Schedule-Alok-radioactive_RC_comments.doc
A: 10/26/2015
This paper describes the removal of Cpn60 from the target protein. Although authors claim that the method can be applied to any protein purification, I tried once and it did not work for me. I am trying again with higher concentrations of ATP. If this works then fine, if not I will think of some different strategies such as including sub denaturating concentrations of Urea (0.5M-2M) in either in lysate or during the wash steps. ArcticExp_Cpn60removal.pdf
RC: Thanks.
RC (10/23): Please provide a brief update sometime today including some commentary on your plan for radioactive experiments (see below).
During the next month, there will also be some additional follow up tasks that we will work on regarding PNAS paper finalization.
AU: 10/23/2015
I used ATP/Mg/Kcl combination to get rid of Cpn60 from final protein. It didn't work. Next week, I will try different concentration of ATP to see if I can get pure Sirt3. There is only one paper I can get so far which deals with this problem in Kinase purification. Hopefully, I will be able to get protein in 1-2 weeks.
RC: Not clear on a few things: I thought the schedule involved doing expression optimization for the first week or so and then over the next couple of weeks work with the ATP method to remove chaperonin.
Has the schedule changed?
AU: No change in schedule. Protein expression, Optimization is pretty much done with arctic express cells. I also have optimized a purification protocol. Only thing remains is to remove Cpn60 from the target protein. I am working on Chaperonin removal aspect (next couple of weeks).
Please see my question below about the expression level changes. I didn't notice whether there was an improvement.
AU: As I mentioned earlier, the expression level in arctic express is not same as in BL21. In BL21, I can see protein expression with IPTG as low as 1uM whereas, in ArcticExpress, the expression starts with 25 uM (with new IPTG solution).
RC: I'm still not quite clear on my question below on this. You spent part of this week or late last week doing expression level optimization in Arctic Express, right? And you had done it before in Arctic Express with different IPTG concentration.
Has it improved? If not, what did you achieve through the latest experiments in IPTG optimization?
AU: When I started purification with Arctic Express cells, I used two IPTG concentration for induction 50 uM and 1 mM. This was based on BL21 expression where I could see expression even at 1uM. Using these two concentrations, the expression level was too low may be about 5-10% of BL21. The good thing was, I could purify the protein at that time. Then I decided to screen different clones to see if I can get a better clone. This time I used 15 clones but none of them were as good as clone 2. I decided to use clone 2 and prepared fresh IPTG. I used clone 2 for IPTG optimization using 1, 5, 25, 50, 100, 200 uM , and 1 mM. I could see protein expression starting from 25 uM IPTG. So yes, the expression level improved significantly with fresh IPTG. Subsequently I used 200uM to express the protein and purify.
RC: How much improvement in expression? It seems this is very difficult to quantify given your reply?
AU: If I have to guess, I would say about 40-50% more from the first arctic express experiment (where I used 50 um and 1 mM IPTG).
As far as radioactive experiment is concerned, I have peptide substrates, and other reagents to start the experiment. I have not ordered radioactive material yet. As it was decided previously, I should have protein first. Also, I would like to mention that the HPLC we have in the lab does not have fraction collector which is necessary to quantify the reaction product. While I purify the protein, I will start looking at the HPLC (with the help of XG, time convenient for both of us) to see if we can test run in the lab.
RC: Regarding our previous discussions, yes we wanted pure protein. Now we (potentially) have a couple of different methods of obtaining it. So those radioactive experiments or at least their planning should not necessarily wait for completion of Arctic Expression purification.
AU: Yes, you are right, we don't need to wait for arctic expressed, cpn60 removed protein, to start the radioactive protein. We can use urea purified protein for initial radioactive experments.
Also, we need to be aware that the implications of using two different methods to purify protein for different papers would be need to be assessed in advance.
AU: Yes, and the only thing I can think of right now is that Arctic expressed protein will have higher specific activity. Plese elaborate "need to be assessed in advance"
RC: I.e., if we start doing radioactive experiments with different purification protocol, we may later find the two batches were not comparable and hence may have conflicting results in the papers.
Thinking about it ahead of time is preferred.
So, we would like to work out a schedule for the radioactive experiments (at whatever detail is possible at this time - I'm sure questions will arise once you start thinking about this more carefully) assuming they will start in about a month or so, and including any preparatory steps before that time.
Regarding HPLC, I'm not sure how this connects with our previous discussion which I thought concluded the current HPLC was sufficient. Regarding looking at the HPLC, please elaborate.
Thanks.
AU: In our previous discussions, July-August, when I was looking at the HPLC quote, I did mention to Sherry that we need fraction collector, meanwhile I started working on Sirt3 purification, she mentioned to me that we can buy fraction collector if they sell it. If not then she can go ahead and buy HPLC as she already got quotes from different vendors.
RC:
Please post your analysis of HPLC fraction collector that can be bought separately.
Please provide the rough schedule of work for the radioactive experiments as well, as discussed above. Questions and issues regarding details will arise and we need to settle those, the earlier the better.
I assume this is underway.
AU: 11/2/2015:
Sherry already placed an order for fraction collector. Expected arrival time is about 2 weeks.
I am in the process of figuring out a protocol to remove Chaperonin from the final protein. The ground work has been done. I need to modify the protocol, use different buffer e.g. Tris-HCl to do the purification and incubate the protein with ATP/Mg/KCl. I have been using Sodium Phosphate. The trace amount of Phosphate may hinder the ATP's action on Cpn60.
I have the basic idea of the radioactive experiment but need time to think and put this in writing. I can hold off the purification for couple of days and finalize the schedule. It will be difficult to do both at the same time because they both need undivided attention. As you suggested earlier, I can use urea method purified protein for assay. Please advise.
RC: You should work on the radioactive protocol for at least 2 days this week and specify which days you will be working on it.
AU: 11-2-2015
I will do it tomorrow and day after tomorrow (11/3 and 11/4).
RC (10/19):
Please note that new hire may start working on chaperonin issue starting in early/mid Nov (initially with you).
You should be ready to start radioactive expts by then because we may need this additional assay results during review of our paper.
You can consider how he might be able to best help (starting by taking over some of the laborious and time consuming tasks).
Please add the preparatory steps required to be able to start radioactive experiments by mid Nov to your draft schedule.
AU: 10-16-2015
Please see the powerpoint (last two) where I have screened several clones for IPTG induction, seems that all are false positive.As of now I think clone 2 is the best. I made fresh IPTG and repeated the titration and this clone can now express the protein with IPTG as low as 200 uM. I am currently doing an experiment to lower than 200 uM. The concentration I am using is 1, 5, 25, 50, 100 and 200 uM IPTG SIRT3-IPTG optimization in Arctic Express cells.pptx.
Once I get hold of the IPTG concentration, I plan to purify the protein with following modifications-
All the steps will be the same as described in the protocol with extra step i.e. incubation of protein with ATP+ MgCl2 in the column.
RC (10/16): You mentioned that we can express the protein at IPTG as low as 200 uM. What about the comparison of expression level at 200uM to that at the IPTG concentration that was used for the previous batch of purification with Arctic Express. Please quantify the difference.
AU (10/16/2015): I never did use 200 uM earlier for Arctic express cells. In the previous batch I used two concentrations to optimize, 50 uM and 1 mM IPTG ( keeping BL21 expression pattern in mind). Since 50 uM induced expression was too low protein then I went on to 1, 3, 5, and 10 mM but the expression was similar in all these cases. Please see updated slide (last in the file) SIRT3-IPTG optimization in Arctic Express cells.pptx
where I used 1, 5, 25, 50, 100, and 200 uM IPTG for this clone. The 50 uM IPTG can induce good amount of protein. I plan to use either 100 or 200 uM for subsequent purification. Any suggestions on amount of IPTG use will be helpful. I am waiting for ATP and MgCl2 to start the experiment.
RC (10/19): I meant to compare the expression level results from whatever IPTG you used in the previous batch to the best results from this batch. I didn't see this comparison in your latest reply above. Please let me know if you can provide this analysis.
Separation of the co-purifying chaperonin-
After loading the column and wash, the protein in the column will be incubated with10 mM MgCl2, 5 mM ATP, and 150 mM KCl at 4 °C for 2 h followed by a 200 ml wash with the respective wash buffer.
RC (10/16): You mentioned that there was some literature that cited this purification method and indicated some of the issues with chaperonin co-elution. You were reviewing this literature vis-a-vis our discussions earlier this week. Please post that literature here with commentary on what you have learned about whether this problem has been solved in the past, and if not, what was the best purity achieved.
Also, please confirm that without further expression level improvement, we would have around 80% purity if this problem was completely solved.
RC (10/24): Was there any such literature found?
AU: 10/14/2015:
Please see attached purification protocol.Sirt3 purification using Arctic Express cells.docx
I will upload the gel picture tomorrow.
Thank you
AU: 9/29/2015: Please see the SDS-PAGE here-SIRT3 expression.pptx
RC: -Please specify when column purification would start for this batch.
AU: I planned to purify the protein on Thursday (this week).
RC: Please also reply to comments on Fri regarding plan for increase of purification scale (e.g., by either using FPLC or combining batches) and the schedule for that, since we would ideally use one batch for initial rate experiments.
AU: I am growing enough cells for two batch purification and I will use manual column to purify them. This will help to establish the purity and yield of the protein. We can determine if we need to combine batches once XG evaluates the activity. If everything goes well, then I will send an email to April and schedule her visit to the lab. I am fairly optimistic that we will be able to purify enough protein for initial experiment.
RC: -It seems SDS increases soluble protein further, but it may not be necessary as long as we get the desired purity without it and efficiently increase the scale of our purifications (above). You previously mentioned we had 15% soluble SIRT3 protein with the original cocktail at room temp, and 80% with b-lactamase. Now we see 40-50% with Artic Blue expression and 0.5% NP-40. What level of soluble protein do you estimate was achieved with 0.4% SDS?
AU: With old BL21 cells, 0.4% SDS gave around 40-50% soluble protein whereas, with Arctic Express the solubility in SDS is >85%. I will avoid SDS for purification.
RC: -It does appear we get more soluble protein at 50uM IPTG. Please clarify what you mean by overall weak expression at 1mM IPTG being beneficial - do you mean the slower induction increasing the amount of soluble protein (with 50uM being better)?
AU: Yes, slower induction increases the solubility. Although 50 uM induce the protein, it may not be sufficient. This can be optimized later once the protocol is established. There are few optimization points (e.g IPTG, number of cells before induction etc) I have in mind but as of now it is unnecessary.
Thank you
Alok
=====
Great! Glad to see that the results are consistent with our expectations, since Arctic Express is a product designed to deal with the problem we were encountering by increasing solubility at low temperature. Given that
we were only seeing 5% increase in soluble protein at low temperature and that other teams were using low temperature cultivation, this was essential to explore.
-Please specify when column purification would start for this batch.
-Per our correspondence on Fri, we would like to see whether this increases our purity to > 90% and simultaneously increases the yield substantially. If we get over 90% purity and 2x yield, then we would need about twice this scale of purification
to do the remaining experiments for this paper (saturating/nonsaturating DHP) in duplicate, three times this scale for triplicate (all intraday). Let us see what purity and yield we achieve - especially if purity is > 90%. Please also reply to comments on Fri regarding plan for increase of purification scale (e.g., by either using FPLC or combining batches) and the schedule for that, since we would ideally use one batch for initial rate experiments.
-It seems SDS increases soluble protein further, but it may not be necessary as long as we get the desired purity without it and efficiently increase the scale of our purifications (above). You previously mentioned we had 15% soluble SIRT3 protein with the original cocktail at room temp, and 80% with b-lactamase. Now we see 40-50% with Artic Blue expression and 0.5% NP-40. What level of soluble protein do you estimate was achieved with 0.4% SDS?
In any case, let's see what we obtain with XG's diluted SDS purifications - this will be helpful for future reference.
-It does appear we get more soluble protein at 50uM IPTG. Please clarify what you mean by overall weak expression at 1mM IPTG being beneficial - do you mean the slower induction increasing the amount of soluble protein (with 50uM being better)?
-Please post our correspondence (including that from Fri) as well as data on the wiki.
Thanks
Raj
====
=====================================================================================
Friday 9/25/2015 meeting update by RC:
To summarize imp points from our meeting today:
- the issue is primarily one of purity, caused by low soluble expression yield of the target protein compared to impurities. column purification does not solve the purity problem due to the very low amount of soluble target protein.
purity obtained with the original cocktail was around 75% (compared to Enzo ~70%).
- hence we cannot solve the problem by increasing the scale of expression/purification - this will not improve purity
- enzo likely encountered the same problem. we both used a SIRT3 sequence that needs to be checked for hydrophobic amino acids.
- low temperature cultivation using the chiller only led to marginal improvement in soluble protein yield (by ~ 5%)
- one reason the chiller may not have helped increase soluble protein expression is that chaperonins were not functioning properly (see below).
- detergent optimization led to an increase in soluble protein level at the lysis stage to 40%, but the detergent also resulted in poor column binding.
upon dilution to 0.1% SDS, the yield of target protein post-column being about the same as that achieved with the original cocktail.
in order to improve, we need to try diluting to even lower [SDS]
- by increasing the level of soluble protein, we address two problems simultaneously, since both the purity and yield will increase. it will be useful in the near
future to get an idea of the effect of a given amount of soluble protein on the ultimate purity. E.g., if we can increase the soluble protein yield to 40%, what purity
would we expect (ignoring the issue of specific activity for now).
- e.g., say we can increase the amount of soluble protein from the original level of 15% to 30%. If this were to increase the purity to above 85%, it would be preferable to Enzo, and it would also double the amount
of protein we can use for experiments.
- once we solve the solubility/purity problem we can relatively easily scale up total amount of protein for experiments. this could be done by either:
a) using FPLC with a larger column - for this, we will contact April asap to check lead time so we are ready when needed
b) combining batches with higher purity
- For example, if we were able to double the amount of protein above, we could do duplicate experiments intraday by either combining two batches or double the purification scale to have enough protein for both 50uM and saturating DHP experiments. Alternatively, if we got high purity but less than double the yield, we could combine more batches or increase the purification scale further to achieve this.
- Besides the above purity issue, if there are any other issues/concerns pertaining to "standardization" of protocol that I should know about, please let me know.
- If we cannot solve the solubility/purity problem shortly, we may consider the use of Enzo batches, combining them to get enough enzyme for all the experiments needed for this paper.
Two conclusions we discussed today were particularly useful:
1) Arctic express cells: I assume this is the transformation Alok was doing early this week, which I was informed about before. I saw this in the new candidate's writeup vis-a-vis low temperature cultivation,
but the mechanism was not completely clear. Having read the attachment it seems that it is quite common for low temperature cultivation (used to increase solubility) to result in misfolding.
The engineered low temp chaperonins are meant to address this. Hence it is definitely a priority to check the increase in solubility from this method.
It seems we should have this data by Mon. You noted this may be done under a couple of different IPTG conditions.
This also raises the question: given that other groups purified at low temperature as well, why did they not use such low temp competent cells? Or did they?
In case of issues, we can also review what the candidate encountered after trying this approach.
2) Intraday duplication of experiments: this was an important suggestion and we should aim to implement it if possible.
Please see above regarding the considerations of yield vis-a-vis intraday duplication.
I will get back to you regarding the rough date by which we need to complete experiments for this paper after the data is in next week.
Regarding the biorxiv, as noted, please let me know if you have any other comments or questions regarding either the paper itself or the
public disclosure discussion we had. As I mentioned, it is useful to get the perspective of people who had not been exposed to preprint servers.
Thanks
Raj
=====================================================================================
AU: 9/28/2015:
http://www.nature.com/ncomms/2013/130902/ncomms3327/full/ncomms3327.html
AU: 7/27/2015
I finished kinetic analysis of WT P99 on Cephalothin. Now, I will start with other two substrate e.g. Ampicillin and Ceftazidime.
AU: 6/22/2015:
Rank order based on preliminary MIC values. If we have computational data, we can compare.Rank order by MIC.doc
AU: 6/18/2015
Starting WT protein purification and activity assay.
Finalizing sequence analysis.
RC (6/19): Ok, when you are ready please post the rank ordered list (by MIC) of clones identified by sequence,
and also indicate which of those are/are not from the originally designed library.
AU: 6/19: I will upload the rank order once I finish sequence analysis. I will look in to the data and update this too.
Regarding WT purification, are you testing the use of FPLC to determine if/how it will be used for subsequent mutant purification?
AU: 6/19: I am using manual purification right now and not using FPLC. I want to get hold of WT enzyme and its batch to batch variation in terms of activity profile. Once I establish that then I might move to AKTA.
Please provide an estimate of when you will be ready for mutant purification from the rank ordered list in case any input is needed for choice of
mutants to purify. Please also indicate the estimated time required per mutant purification and activity assay when available.
AU: 6/19: I am not there yet for mutant purification, may be two week from now, I will be able to tell you exactly how much time needed to purify a mutant. Some time it is hard to estimate the time for mutants as they behave differently.
RC (6/19): Please see attached for comments and questions on the screening discussion report. Overall the report looks good (will need some revision of the language, including writing in the form of a paper rather than internal discussion). Priority are the content revisions mentioned.
Please add revision of this doc to your task list for the next couple of weeks.
AU: 6/19: I will update the write up as per the feedback.
Screening method discussion.docx
AU: 5/29/2015:
1: I finished MIC determination of all the clones I had and waiting for the sequencing data back from the Genewiz for last plate. Once I get sequencing data, I will annotate and align all of them to find out what mutation they have.
2: I will finish labeling all the mutants by name. As of now they are named as clone name..
3: I will finish plasmid isolation from plate 3
4: Collect purification, activity assay buffer information from the literature as different research group have used different buffer.
AU: 5/15/2015:
I finished MIC determination for lib2 plate4. Need to repeat 6 samples.
Started plasmid isolation and by the mid of next week, I will send DNA for sequencing.
I need to re-sequence few clones from earlier plates, I will try to finish that too then finalize the data.
-----
RC: Please post your outline on comparison of our screening methods with others (hts) to wiki and update as you develop it further.
AU: The b-lactamase library screening methodology consists of two part-
Part 1: We screen clones based on the expression of protein. All of the b-lactamase papers, I have read so far, used dot blot (western blot) to identify good clone. Since we do not have western blot and X-ray developer, I am doing this step using protein gels.
Part 2: This is were we screen for the activity of the enzyme, in vivo, (growth assay/MIC determination). There are two valid methods available to perform this screening-
A) on solid agar plate
B) in liquid media; I am using this method.
I am not sure if this answers your concern, if not please expand. I am working on to finish literature task, but wanted to finish this first, and update once I am done.
AU: 5/7/2015:
MIC determination for plate 4 is underway, completed Amp and Cephalothin. Need to do some repeat which will be done next week.
By the end of next week, I will send out samples for sequencing.
AU: 4/23/2015:
I will start screening plate 4 of the library.
There was some problem in sequencing plate 3. I am in touch with Genewiz to find out what the problem is. To this end I isolated plasmids from 10 samples and send it out for re-sequencing. I will know next week and update wiki accordingly.
AU: 4/9/2015:
MIC determination for Lib2 plate 3 will be completed today (results tomorrow). Start preparing for plasmid isolation and send it for sequencing, most probably on Wednesday.
Here is the rank order I prepared for mutants Rank order by MIC.doc I have and this list will grow as I get more mutants. Please see the document for rank explanation and please feedback for better ranking system.
AU: 4/1/2015:
Finished protein expression analysis of Library 2, Plate 3. Total 52 clones are positive for IPTG induced protein expression.
Following clones are +ve, clones are listed in 96 well format:
A1, A3, A5, A6, A7, A8, A9, A11
B5, B7, B8, B10, B11, B12
C2, C3, C4, C6, C7, C9, C10, C11
D1, D2, D4, D5, D6, D7, D8, D9, D11
E1, E3, E5, E7, E9, E10, E11
F1, F2, F4, F5, F10, F11
G3, G4, G6, G8, G9, G10, G11, G12
Now I am preparing for MIC determination of these clones and start from 4/2/2015.
AU: 3/27/2015:
I am step 3, running gel to pre-screen potential clones from the plate 3. By the end of Tuesday I will finish protein gel and be ready for next step. I on the schedule for lab work.
1: Prepare media etc to revive the cells.
2: Protein induction and harvesting
3: Run protein gels
4: Plasmid isolation
5: MIC determination.
AU 3/24/2015:
Task Schedule: Task Schedule-Alok.doc
AU: 4-17-2015: I am slightly behind my schedule in literature part. I will take couple of more days to finish this. thanks
AU 3/23/2015:
This week, I will start lab work, plate 3 of the library screening which will continue next week. I will update status on Thursday/Friday.
1: Prepare media etc to revive the cells.
2: Protein induction and harvesting
3: Run protein gels
4: Plasmid isolation
5: MIC determination.
Simultaneously I am collecting and reading papers related to the project specifically experimental/computational library screening and subsequent refinement of the model. I will update status of this too by the end of the week (Thursday or Friday) along with other updates.
AU 3/13/15:
Right now I am working on three different tasks with priorities as follows.
1: Protein purification optimization for P99 WT on AKTA system.
2: Literature search and working on to answer Dr. Chakrabarti's queries related to b-lactamase project.
3: Finish sequence analysis and finalize the mutant table.
Since all three assignments are different and they can not be done simultaneously. But
So far I am halfway through for each of the tasks. I need 4-5 weeks to finish #1-3 (assuming no technical difficulties in #1). By the end of Task #1, I will be able post a method to purify P99-WT and may be able to compare with manual purification process (although not necessary, this will be more relevant to Sirtuin protein purification if XG wants to compare activities). Task 2 and 3 will be completed during break time, whenever AKTA is running and break is more that 30 min. But finally I will need extra 1 week** to finalize all three.
RC: I would like more fine grained detail on the tasks; please post the specific tasks from the list provided and comment on the order/schedule, and current status of each.
AU: Finished one run on AKTA. System is ready to work for His-tag protein but more optimization is required for fine tuning. We (Me and XG are planing to send data to GE and get some clarification which will help in future). Next week I will resume B-Lactamase work.
Regarding efficiency of screening, there are many journal reports that characterize the maximum library size that can be effectively screened in several months with various methods.
Some of these claim orders of magnitude greater library sizes than those we are sampling experimentally (e.g, 10^5-10^7 mutants). These often rely on analytical methods capable of rapid readout in various types of plate-based assays. We should break down the time required for each step and explain why our screening efficiencies are different. In the Nature methods/protocols, I did not notice that it mentioned the library size that could be screened.
Please also provide the rate (e.g., number of unique mutants/month) of screening we have achieved so far.
Of course, our goal is to screen more focused libraries generated by computation (effective size of library searched computationally is much bigger), so the studies are not directly comparable. But we may still be limited by the speed of our experimental screening.
AU: I started in September 2014 and as of today about 6 month, I have total 37 unique mutants of the library gives me 6 mutant/month. Total 37 unique mutants After 3 plates.doc
I have finished three 96 plates out of six.
AU:
These are the steps I have to do to get mutant information in b-lactamase library. There are several factors which affect the efficiency. I am still searching for a paper which directly compares specific method evaluating screening efficiency.
A review article by Leemhuis et al., 2009 (provided by Dr. Chakrabarti) lists general strategies to screen enzyme libraries, and touches some of the pros and cons of the methods used in screening enzyme libraries, mostly E-S reaction and product visualization. The review paper states that “Microtiter plate screenings thus enable the use of various analytical tools and whilst they offer a great dynamic range, their screening capacity is usually limited to less than 10^4 variants per day”. This statement is context dependent and related to specific enzyme assay (Histone Acetylation or similar ELISA assays).
Prescreening: 11-12 days to process 96 samples:
To see if clones express good protein (b-lactamase). In our case, this has two steps as opposed to one step frequently cited in the literature. Most of the literature I read so far, they used TEM1 promotor in their plasmid. This is a constitutive promotor as contrast to our inducible promotor. The best thing about TEM1 promotor is that they don’t have to be induced for protein production, and can directly go to immunoblotting step (e.g. colony dot blot). The immunoblotting experiment can itself be done on high-throughput scale, which saves lot of time. Since we don’t have system to do Western Blot, I am prescreening the mutants by running SDS-PAGE. While my method is as valid as other described above, it may have some limitations e.g. slightly time consuming. At the end, I get about 40-60 (out of 96 samples) samples expressing protein. The next step is to prepare plasmid for sequencing.
Sequencing and analysis: 7-9 days (80-120 samples (each plasmid is sequenced twice):
Each plasmid gets sequenced twice so 40-60 plasmids becomes 80-120 sequencing samples.
Once I get data about protein expression, then I grow cells for plasmid isolation. It takes almost whole day to isolate plasmids from 24 samples. I generally send my samples on Thursday so that I receive data by either Monday or Tuesday.
This step could have been faster with better equipment and reagents e.g. high speed centrifuge which have adapter for 96 well plates and compatible plasmid isolation kit (I have never used this method), nonetheless, I try to be on schedule and finish plasmid isolation so I can send it out on or before respective Friday. Once I get sequence data back then I clean, annotate, translate into protein, and then align with WT P99 to get mutant information (is there way to automate this?). To some extent, this step is semi high-throughput. Some have done sequencing in 348 well formats depending upon number of mutants the plan to screen.
Final screening (MIC value) 8-10 days for all three antibiotics with repeat experiments if needed : As soon as I send plasmids out for sequencing, I start preparing for this step. I am doing this step in 96 well format using Broth dilution method with three different antibiotics. In liquid culture, 96 well plates are well suited as a clear conclusion can be drawn regarding bacterial growth. I choose liquid culture because we have inducible promotor in the plasmid. They are easier to grow/induce as compared to Agar plates with IPTG, and have high reproducibility as compared to Agar plate method. Both Methods are equally tedious, and time consuming.
RC (3-20): Information above will be useful. As we discussed, please post on the wiki the schedule and status of all (10 to 15) tasks listed in our email correspondence today (see comment above regarding fine grained detail on task list).
AU (3/20): I am still searching for the relevant papers to answer some of the questions. This word doc has most of the answer (some overlapping of posted above) b-lactamase wiki response.doc . Later, if I get a general sense, then I will update same document today. If not then I will do it on Monday.
RC: I am referring to posting the list of 10-15 tasks from my email to the wiki, indicating the order in which they will be worked on, the status of each, and the rough schedule of work.
The answers to the questions can come later as you read the papers (only some of those tasks pertain to literature).
As noted a couple of weeks ago if posted at the outset this will help facilitate communication on the work as it progresses.
Some were described verbally but it is desirable to have them posted here.
I noted that they are in the word doc above; thanks. I assume you will be updating this document with the planned order of future work? It is desirable in the future to list the order and rough schedule at the outset of the work as noted.
AU: Yes the word doc (b-lactamase wiki response.doc) has all, the question and most of the answers. I will update literature part just need more time as questions are very specific. Do you want me to finish this first? otherwise I was planning to start working screening rest of the library fro Monday. Thanks.
RC: Not all the answers, but the order, status and schedule of these 10-15 tasks should be posted first since the timeliness of posting the schedule is important (it should be done in advance of the work whenever possible). As noted I am not looking for the final answers at this time.
AU: please see new post. Thank you.
AU 3/5/2015:
I am working on 4 assignment.
1: AKTA optimization
2: Literature search and working on to answer Dr. Chakrabarti's queries related to b-lactamase project.
3: Finalizing sequence data.
RC: Alok, please post the order in which you are working on the tasks listed in the recently emailed task list.
AU: Since I am waiting for some part for AKTA equipment, I am mostly working on your email questions. May be next week, once I receive part, the priority will be as listed above.
I already talk to her regarding AKTA use and we agreed to meet and discuss. A AKTA purification protocol will be similar to the Manual protein purification protocol. I will post a general protocol for purification soon but a final version will be uploaded when it is ready.
If it is helpful, you can arrange a meeting with XG to discuss and plan the schedule.
Also, I believe one of the tasks was a protocol for AKTA purification. You can post a rough protocol that then can be optimized
as indicated above. This will also be useful to XG.
AU 3/2/2015:
Continue last week's work.
Received Lib2-plate2 sequencing data from Genewiz. I will devote some time to analyze the sequences while waiting AKTA accessory.
Grow WT P99 large scale to purify from AKTA.
AU 2/27/2015:
Finalizing sequencing data for Lib2-plate2.
Working on AKTA method development protocol.
AU 2/19/2015:
Finished MIC determination on all three antibiotics, Amp, Cephalothin, and Ceftazidime for Lib2-Plate2 clones.
I the process of isolating plasmids to send it out for sequencing on Tuesday next week.
I will start working on to optimization and method development on AKTA for protein purification and purify P99 WT using His-Tag column. This may take 1-2 wk or more.
Once I develop purification method on AKTA, I will start working on B-Lactamase project (Lib2-Plate3).
AU 2/12/2015:
Completed MIC determination Lib2-Plate2 clones.
Some repeat experiment will be done next week.
Plasmid isolation tomorrow and send samples next week.
Start with Lib2-Plate3 protein expression.
AU 2/5/2015:
This week & possibly go in to next week:
1: Finishing up MIC determination (three antibiotics) of IPTG +ve clones of Lib2-Plate2.
2: Will isolate DNA for sequencing by the end of next week and send it out for sequencing.
3: Prepare for IPTG screening of Lib2-plate3.
4: Upload the data on relevant section by end of the next week.
5: Literature survey related to this project.
AU 2/3/2015:
Finish MIC determination of Lib2, plate 2 clones (Three antibiotics).
Prepare plasmids for sequencing (lib2 plate2).
Start IPTG expression for Lib2, plate 3, 4, and 5.
AU1/29/2015:
Finished Library2 plate 1 screening.
So far got 28 of 128 mutants of the library
Finished IPTG expression of Library 2 plate 2 screening
Next week I will start MIC values and plasmid isolation.
AU 1/27/2015:
Continue previous week's work.
Finalize Lib2 plate 1 mutant data.
AU 1/22/2015:
Finished IPTG expression for 96 clones (Lib2 Plate 2).
I will start MIC determination and plasmid isolation for these clones so that I can send them for sequencing.
Analyzing sequencing data for lib2 plate 1 clones. I will update result section as data gets final.
AU 1/19/2015:
Continue previous week's work to finish, start MIC determination (Lib2, plate 2)
Sending out samples for sequencing (Lib2, plate 1).
AU 1/15/2015:
Analyzing sequence data, clean up, sequence alignment, and mutant identification (Lib2 plate1)
Running protein gels to assess protein expression (Lib 2 plate 2).
AU 1/12/2015:
Sending out 49 clones for sequencing (of Lib2, plate 1).
Start protein expression analysis for Lib2, Plate 2 clones.
Please see Beta-Lactamase Redesign for some new results.
AU 1/9/2015:
I will update on Monday..Thanks
AU 12/29:
Finishing up repeat experiment to calculate Cephalothin MIC values.
Finishing miniprep to send plasmid out for sequencing.
AU 12/22:
I have updated
Beta-lactamase Redesign section!
I am currently making sense of sequencing data (annotation etc)
Repeat some of MIC data for library 2, plate one. more updates on Tuesday.
AU 12/18:
Updated Beta-lactamase Redesign section.
As far as experiments are concerned, it is same as previous week and repeat some of MIC data for Library 2, plate 1.
AU 12/15:
Sending out plasmid for sequencing and finish plasmid preparation for plate 1.
Finish MIC values for Ceftazidime.
Repeat some of Cephalothin MIC for plate 1.
Once plate 1 is complete i.e. IPTG expression, MIC on three different antibiotics, and sequencing, then I will move to plate 2.
AU 12/11:
Finished preliminary screening (IPTG expression) of 96 clones, their MIC values with Amp and Cephalothin.
Miniprep IPTG (+) clones for sequencing, make glycerol stocks.
Optimizing amount of Ceftazidime to determine MIC value for WT.
Started IPTG expression for another 96 clones for subsequent MIC value determination.
Literature survey to get protein purification/assay protocol for WT P99.
AU 12/1:
Finished on set of IPTG expression, sequencing and Approx MIC determination see data file in relevant section.
Adding one more antibiotic for MIC determination, Ceftazidime, as per discussion.
IPTG screening, MIC value determination is in progress, Sequencing will be done at the end of each plate (after 96 colonies screened)
AU 11/24:
Finished one set of sequencing, and have MIC data of mutants. I will upload the entire file in relevant section, possibly by the EOD.
Focus more on WT enzyme assay.
Continue IPTG screening and growth assay as planned previously.
AU 11/17:
I am purifying WT b-lactamase to start activity assay etc.
Continue growth assay as planned previously, I am not in the lab 19th and 20th.
AU 11/14:
Continue screening clones in the library by IPTG.
Continue growth assay of the clones already screened with IPTG, just waiting for Ceftazidime.
Will start mini prep for clones so that they can be sequenced.
(this 3 points will be repeated in my posting until I complete screening of ~500 clones).
AU 11/10:
Analyzing dna sequences after sequencing. preparing final data for this set. I will update relevant section of wiki once I complete it.
Continue screening clones in the library by IPTG.
Continue growth assay of some of the clones already screened with IPTG.
AU 10/31:
Continue screening clones in the library by IPTG.
Continue growth assay of some of the clones already screened with IPTG.
Started mini prep for clones so that they can be sequenced.
Reading assignments related to SBIR proposal.
AU 10/27:
Continue screening and growth assay along with mini prep.
AU 10/24:
Continue screening clones in the library by IPTG.
Continue growth assay of some of the clones already screened with IPTG.
Started mini prep for clones so that they can be sequenced.
AU 10/23:
I will update wiki by end of the day. Thanks
AU 10/20:
Two week, I will be doing screening potential library for protein expression by IPTG.
Finish growth assay of some of the clones already screened for IPTG.
AU 10/16:
I believe, I have optimized growth assay in liquid culture. I did experiment two times, pretty consistent results.
I have purified WT P99 and I think the protein is more than 95% pure.
I would like to discuss agenda for next week in the group meeting, unless something comes up, my focus for next few week will be to continue with growth assay and start screening clones (around 500) for IPTG induced expression.
AU 10/13:
This week, optimization of growth assay and WT protein purification.
I will try to screen more IPTG inducible clones for the library.
AU 10/9:
I am pretty much on schedule.
I finished rest of protein expression analysis for potential library (total 88, will update relevant wiki section). I need a discussion how I am proceeding further with these data in for growth assay screen. Since vector we are using (pET) is IPTG inducible, I can not follow Cornish (2003) exactly. I have to come up with new and valid protocol for screening assay (for MIC determination). I have three methods, please see attached file.Activity screen in liquid.docx
Right now I am at method 1 (just for WT) and I will know tomorrow if it worked. Please look at the basic protocol I made (which can be modified as I get data and Dr. Chakrabarti's input).
If my protocol works then fine, I will start screening (I haven't decided time line yet). And if it doesn't work then we have to think about targeted approach e.g. sequence clones first, then proceed further.
I have library in both DH5a and BL21 cells and we can decide where to start after screening experiment. WT protein purification is pushed to next week as protease inhibitor arrived today. So next week will be continuation of this week's work, possibly I will have purified protein in hand and plan/do some initial assay experiments. At this stage, this data can be feedback us for screening.
WT protein purification pushed to next week as protease inhibitor arrived late today.
AU 10/6:
I will finish IPTG induced protein expression by tomorrow.
Most of the time, this week, I will focus on optimization of WT growth assay and WT protein purification.
AU 10/02:
16 more clones to be analysed for IPTG induced protein expression in the existing library.
Waiting for BL21 competent cells so that I can do transformation to cover all possible mutants in the library (Need to do fusion PCR, restriction digestion and ligation)
Next week will focus on WT growth assay, WT protein purification etc..
AU 9/29:
Continue protein expression analysis of rest of the potential mutants in the library.
Repeat transformation in BL21 cells
WT growth assay combining liquid and plate culture
If time permits, start WT protein purification
AU 9/22:
Confirm protein expression in mutants before I draw any conclusion.
Ligation and transformation in BL21, fusion PCR
Order IMAC resin and colum for WT protein purification.
AU 9/18:
Analyzing sequencing data, annotate and upload on wiki as soon as its ready.
Made glycerol stocks of WT P99 and mutant clones AU1, AU2, AU3, AU4 in BL21
Did initial protein expression analysis, using two different concentrations of IPTG (0.5 mM and 2 mM) for WT, AU1-4
Since I do not have a standard for protein, it is hard to interpret, I will upload gel pic on wiki.
So far I am on schedule, and will update on Friday if I am ready to order resin for protein purification.
Things are on schedule for next week: -protein expression analysis, mutant library ligation and transformation in BL21 (may need to do fusion PCR).
AU 9/15:
Sequencing data is back. I will analyse, annotate and upload on wiki.
Sequencing looks clean to some extent, I will send more plasmids for sequencing.
Transform AU1, AU2, AU3, AU4 in BL21
Along with mutant plasmid isolation, I will optimize P99 WT protein expression.
I will add more task in this section depending upon outcome of above points and update.
AU 9/11:
- I am on my schedule, most of work I proposed last week completed.
- continue doing miniprep to get maximum numbers of potential mutants of the library.
- growing WT P99 in BL21 for growth assay
- later in the week, evaluate protein expression analysis, optimize IPTG concentration for optimal protein expression
- Grow WT P99 plasmids, send it for sequencing
- Make LB-Kan plates
- Library ligation, transformation, prepare for miniprep, send it for sequencing
- Do PCR to generate more insert library
•Make mutant library using Chaoran generated fragment [1 wk]
•Vector preparation [2-3 wk]
ØOnce I get WT P99 sequence back
ØMidi prep pET 26b(+) vector
ØRE digestion, gel purification
•Once Pfu Turbo comes in [2-3 wk]
ØPCR to make individual fragments, PCR clean up
ØMake full length insert library, Gel purification/PCR clean up
ØLigate following CJ’s protocol, Transformation
AU (9/4):
I was unable to locate plasmid "WT P99 CJ01" in CJ's reagent box but successfully transformed "WT P99 V" plasmid in DH5a. I assumed
"WT P99 V" is the WT P99 clone.
I included a mutant P99 Y241A as a control as transformation didn't work last time.
Transformation worked for both.
Today, I will grow 3 WT P99 clones and do mini prep tomorrow.
I will design primers and send it out for synthesis. Later these primers will be used to sequence at least 3 WT P99 clone to evaluate gene sequence.
Meanwhile I will make glycerol stock of WT P99 clones and store them at -80.
AU (8/29): XG, please let me know where can I find Pfu turbo polymerase and Kanamycin. Thank you.
AU (8/27): Thank you XG for feedback.
Why DH5α: This is a cloning strain. For plasmid prep, it gives much better yield as compared to BL21 which is expression E.Coli. While reading Dr. Jing's note book, I found that she had very low yield of plasmid preparation and I suspect that is the reason.
I can do midi or maxi prep whichever is available in the lab. The idea is to get good amount od plasmid so that I don't have to prepare multiple times.
AU: 8/26/14
Hello All,
While reading Dr. Jing's notebook, I am preparing note to myself and would like to proceed as described below. Any feedback/suggestions are much appreciated.
Thank you
Alok
Things to do (in next two week):
- Need to locate WT P99 CJ01 and CJ02 plasmids or bacteria containing plasmid.
- Transform in DH5α, Make a glycerol stock.
- Do maxi prep; store DNA at -20OC for future use, mostly for backbone.
- Design and synthesize new internal primers for sequencing.
- Send WT P99 plasmid DNA for sequencing with new internal primers (at least 2 primers).
- I assume mutant plasmids are confirmed by sequencing and validated.
- Since assembled fragments are mixture of mutants, they cannot be sequenced. I need to send fragment 3 alone (just WT, if possible) for sequencing using Reverse primer (CJ22, made by Dr. Jing) to see if missing “G” is there are not. If fragment 3 WT alone is not available, then see next step.
- If everything is fine in sequencing, I can quickly try to generate both vector and insert to ligate with new ligase enzyme. Once I get colonies, I will send few samples for sequencing to confirm integrity of fragments.
- I am not sure if Dr. Jing has purified WT P99 protein and did activity assay etc. So to this end, I am thinking of using WT P99 and do growth assay etc., and purify protein so that I can start with activity assay. It will save time later when I am ready with library.
- Major discussion point will be if IPTG inducible pET 26b (+) is a good choice especially for screening (reproducibility).